





1. INTRODUCTION
Achalasia is a rare esophageal disorder. Although etiology of the disease is still largely unknown, its resultant abnormalities have been well understood which include loss of esophageal peristalsis and relaxation failure of the lower esophageal sphincter [1]. Current pathophysiologic knowledge of achalasia suggests the important involvement of genetic predisposition [2-4]. However, there are few studies on familial achalasia in the literature. Most of the case reports are small with the average number of patients from 2 to 4, mostly in siblings than in members of different familial generations.
To the best of our knowledge, there are only very few case reports on familial achalasia with classical autosomal inherence pattern reported in Europe and the United States [5-7]. We hereby report a Vietnamese family with 10 affected members in three generations. This is the first such a case of familial achalasia with classical autosomal inherence pattern reported in Vietnam.
2. CASE REPORT
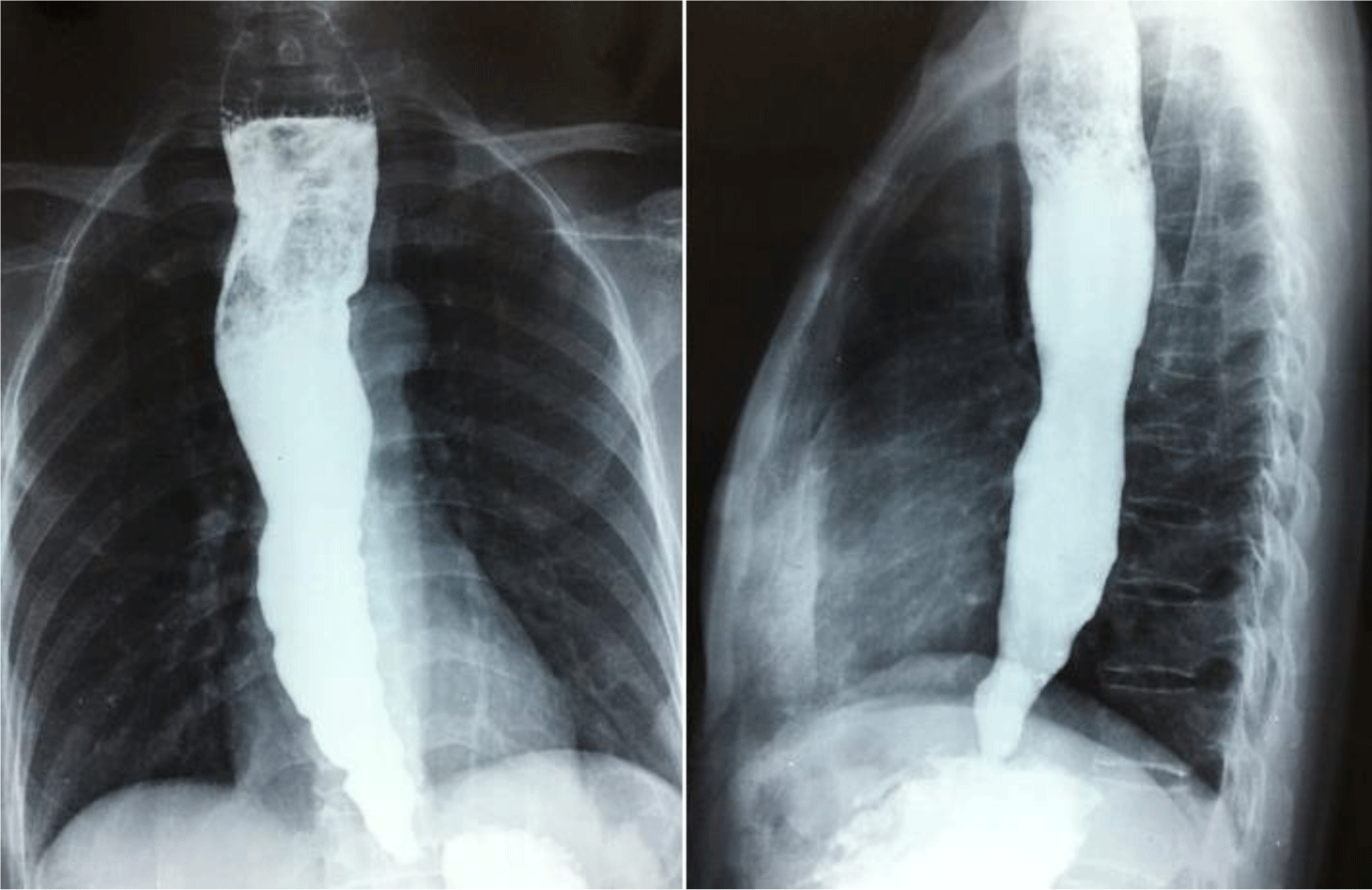
A 50-year-old female patient was referred to the University Medical Center, Hochiminh City, Vietnam because of worsening dysphagia. She had dysphagia for 20 years, initially for solid foods but subsequently for liquids. Upper gastrointestinal endoscopy showed significantly dilated esophagus with retained secretions. A high pressure zone was felt and a “given” feeling was experienced as the scope passed the gastroesophageal junction. There was no true stricture or neoplasm. Achalasia was suspected and upper gastrointestinal series were performed which confirmed the diagnosis (Figure 1). Endoscopic pneumatic balloon dilatation was performed successfully.
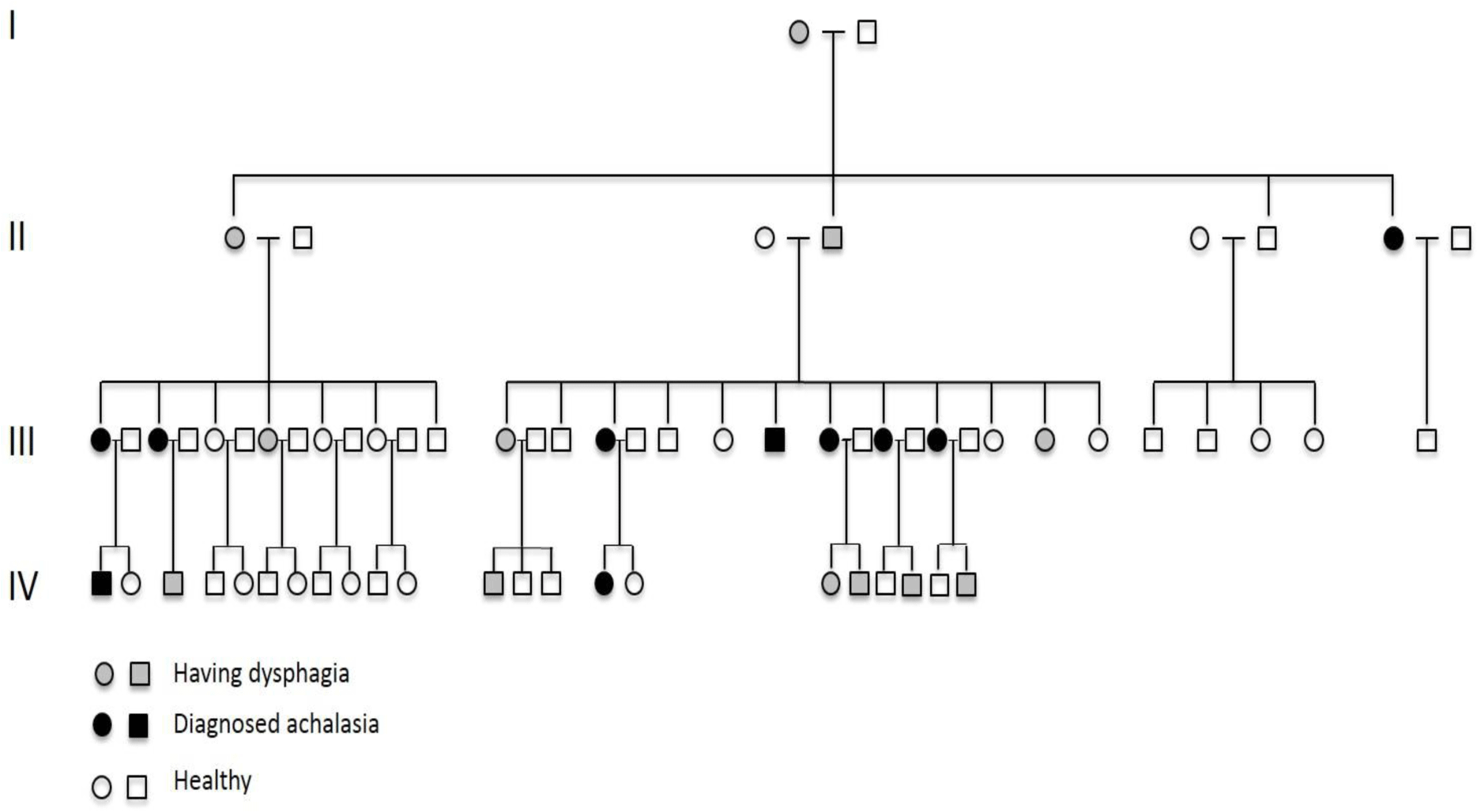
This is a four-generation family. There are ten patients (two male, 8 female) belonging to three different generations with confirmed achalasia (Figure 2). The diagnosis was made based on dysphagia, typical esophagram of achalasia and upper gastrointestinal endoscopy (to exclude other organic causes of dysphagia) at two tertiary hospitals in Hochiminh City, Vietnam (Nguyen-Tri-Phuong hospital and University Medical Center). Esophageal manometry was not performed as the equipment was not available in Vietnam. Because of different management strategies at the two hospitals, six patients admitted to Nguyen-Tri-Phuong hospital were performed Heller’s myotomy surgery while the other four patients admitted to University Medical Center were performed endoscopic pneumatic balloon dilatation. One patient with initially unsuccessful balloon dilatation was managed by myotomy surgery. In addition, there are 12 members of the family belonging to 4 different generations suffering from dysphagia. All of the family members who have been diagnosed with achalasia or suffered from dysphagia but not yet been confirmed with achalasia reported that they firstly had dysphagia at the age of 15 to 25 (Table 1). There has been no esophageal carcinoma diagnosed in this family.
3. DISCUSSION
To diagnose achalasia in the early stage of the disease, the guideline of the American College of Gastroenterology recommended to use esophageal manometry as the primary examination; and barium esophagram and upper gastrointestinal endoscopy were considered as complementary tests [8]. However, this guideline also addressed that the role of manometry is supportive to confirm the diagnosis in patients with classic endoscopic and esophagram findings. Although esophageal motility testing was not available in Vietnam at the time these patients were managed, patients with achalasia diagnosis in this family were in late stages with typical esophagram findings (esophageal dilation and peristalsis, narrow gastroesophageal junction with poor emptying of barium) and endoscopic findings (esophageal dilatation, narrow gastro-esophageal junction with no evidence of cardiac tumor) and, therefore, the definite diagnosis of achalasia could be reliably established. Affected members in the fourth generation of this family were diagnosed and received treatment at younger ages compared with those in the older generations as a consequence of the awareness of this disease.
Most of the cases of familial achalasia presented in literature showed an autosomal recessive pattern of inherence. A large prospective cohort study over the last 24 years in Nigeria recently reported 18 families with familial achalasia [9]. All patients in these families were observed in siblings, and consanguineous marriages were common among parents of these patients. There were very few case reports on classical autosomal inherence pattern in the literature. One case is a Southern American family with six affected members in three generations. The beginning age with symptoms was about 22 – 25, similar to patients in our case report. Two members finally died because of esophageal carcinoma in their late seventies and eighties [6]. Another case is an Italian family with five affected members in three generations. The beginning age with symptoms was not mentioned and there was no esophageal carcinoma occurred during the follow-up period [5]. The number of members with confirmed achalasia in our case report was the largest among the three case reports. In addition, 12 members belonging to four generations suffer from mild dysphagia. The subjects have not consulted or already consulted but have not been confirmed with achalasia. If motility testing were available, there would have been more cases with achalasia diagnosed among these subjects. The pedigree of this family clearly showed a vertical transmission of the disease and strongly suggested the autosomal dominant pattern of inherence (Figure 1).
In genetically predisposed subjects, it is suggested that environmental factors may trigger an autoimmune antibody response against myenteric plexus of the esophagus [10]. In fact, a recent large case-control study reported that patients are more often affected by viral infections before achalasia onset, most significantly for varicella-zoster virus infections [11]. In addition, this study also showed that achalasia can also be triggered by pregnancies in female HLA-DQß1 insertion carriers. These findings help to explain the dysphagia onset around the age of 15 – 25 years and the female predominance among affected members in our presented family. Recent studies reported that allergic and autoimmune disorders are comorbid disease conditions in patients with achalasia [12, 13]. However, we have not identified any significant comorbid conditions in members of this family. In addition, no cases with esophageal cancer have been diagnosed in this family until now. However, patients diagnosed with achalasia in our case report are still at much younger ages compared to those with esophageal carcinoma in the first case report [6]. Therefore, the development of this malignancy needs to be further followed-up.
In conclusion, we report for the first time familial achalasia with autosomal dominant pattern of inherence in Vietnam and review the literature. Future genetic study is required to identify the causative gene.