
1. INTRODUCTION
Mycophenolic acid (MPA) is an active metabolite of mycophenolate mofetil and mycophenolate sodium which have been widely prescribed for the purpose of preventing organ transplant rejection. MPA has a non-competitive and selective inhibitory effect on the inosine 5’-monophosphate dehydrogenase (IMPDH), which is required for T and B lymphocyte proliferation. However, MPA has a narrow therapeutic window, which can lead to risks of either side effects for overdose of the MPA and a graft in under-dosing MPA treatment. Furthermore, variation between and within patients can affect the therapeutic results [1-3]. Although patients received the same MPA doses, they reached different therapeutic efficacies. Pharmacokinetic monitoring of the MPA in transplant patients may better prevent allograft rejection and reduce the toxicity of the MPA. Determination of MPA levels in plasma helps to adjust MPA dosing.
Currently, many quantitative methods have been developed to determine MPA plasma concentrations, where high-performance liquid chromatography (HPLC) is a chromatographic technique used to separate complex mixtures of molecules in biological matrix [4-7]. Previous studies showed that the enzyme multiplied immunoassay technique (EMIT) method is less specific than the HPLC due to metabolites cross-reactivity, displays therefore slightly higher concentrations than HPLC. The internal standards (IS) are often used in chromatography to correct changes due to the analytical process that contribute to improve the accuracy of quantitative analysis. The objective of this project is to develop and validate a HPLC-PDA (photodiode array) method using an IS for quantitative determination of the plasma MPA concentrations.
2. MATERIALS AND METHOD
Sample purification was treated using OASIS HLB cartridge 1 mL (Waters, USA). LiChroCART®125-4 (C18 reversed-phase column) was purchased from Merck (Germany). Liquid chromatography was performed on HPLC system 2695 XE (Waters) equipped with a PDA detector.
MPA was purchased from Toronto Research Chemicals Inc. Methanol (MeOH) and acetonitrile (ACN) were obtained from Merck. Visnadine and triethylammonium phosphate (TEAP) buffer 1 M pH 3) were obtained from Sigma-Aldrich. Phosphoric acid was obtained from VWR BDH Prolabo. Human plasma samples for analytical development were obtained from Cho Ray Hospital in Ho Chi Minh City, Vietnam. The local Ethics Committee has approved the blood sampling for this purpose.
Solutions of 4% phosphoric acid and 5% methanol were prepared by diluting the concentrated solutions with double distilled water. TEAP buffer 0.25 μM (pH 3) was obtained by diluting 25 mL of buffer 1 M (pH 3) in 975 mL of double distilled water.
The stock solutions were separate solutions of 1 mg / mL visnadine and 1 mg / mL MPA in absolute methanol. Working solutions of 400μg/mL visnadine and 100μg/mL MPA were prepared by diluting the stock solutions with methanol.
MPA calibrations and quality control (QC) samples were prepared separately by adding 25 μL of visnadine (400 μg/mL) as internal standard (IS) into 300 μL of plasma fortified appropriate amounts of MPA to obtain concentration of 0.25, 0.5, 2, 5, 7, 10 μg/mL for calibrations and 0.75, 4, 8 μg/mL for QC samples, respectively. These samples were then processed similarly to the real samples. For samples obtained from patients under MMF therapy, only 25 μL of 400 μg/mL visnadine were added to 300 μL of plasma.
300 μl of plasma, 25 μl of 400 μg/mL visnadine and 325 μL of 4% H3PO4 were added to a 2 mL Eppendorf tube. Plasma samples were vortexed for 30 min at room temperature (RT) and then centrifuged at 14,000 xg for 10 min to remove protein precipitate. 650 μL of supernatant were collected and placed in Osis HLB 1cc cartridge (Waters). Next, this cartridge was washed either with 1 mL of 5% methanol or 10% methanol. Then, MPA and internal standard (IS) simultaneous extraction was examined by adding either 500 μL of pure methanol or 50% methanol. The solution eluted from cartridge was collected into a 2 mL Eppendorf and evaporated to dryness at 65 °C. The dry fraction was vortexed in 50 μL of methanol for 1 min. After centrifuging at 14,000 xg for 5 minutes at room temperature, 10 μL of the supernatant was transferred to the vial for MPA measurement.
10 μL of sample extract was injected onto LiChroCART®125-4 at 43 °C on a Waters 2695 XE system. The samples were kept at 4 °C in autosampler. The mobile phase was a mixture of solvents A and B maintained at a flow rate of 1 mL/min. Solvent A was acetonitrile, and solvent B was TEAP buffer (pH 3). Over 0–0.5 min, solvent A remained at 15% and linearly increased from 15 to 40% for 2 min, and continuously increased from 40 to 62% for 4 min and kept for 4.5 min, then returned to 15% over 0.5 min and kept for 3.5 min before the next injection. Total chromatographic runtime was 15.0 min.
The HPLC chromatographic method in this study was validated according to the guidelines of the US Food and Drug Administration (FDA).8 Data were processed and analyzed with JMP 10 software (JMP software; SAS Institute, Cary, NC). Statistical analysis was considered significant if p-value was <0.05.
Selectivity was assessed with 6 blank plasma samples. MPA and IS were spiked in blank plasma samples to differentiate the analytes from other components in the matrix. Absence of interfering components is accepted where the response is less than 15% of QC levels of MPA and 5% for IS [6].
The linearity of the method was evaluated on 5 calibrators in replicate of 3 in 3 runs. The coefficients of determination R2 should be > 0.99. The coefficient of variation (CV) should be 20% for the lower quantitative limit (LLOQ) and should not exceed 15% at other concentrations. Calibration curves were based on the ratios of MPA/visnadine peak area versus MPA concentrations.
The accuracy and precision were validated on QC samples at 3 levels (0.75, 4, 8 μg/mL) in replicate of five. Intra and inter-assay accuracies were calculated from the differences between the nominal and the observed concentrations. The within-run CV% value of precision and accuracy should be 15% for the QC levels and 20 % for the LLOQ.
Recovery of MPA in plasma was calculated by comparing MPA extracted from QC samples at 0.75, 4, 8 μg/mL with standard MPA in MeOH.
LLOQ is a concentration with accuracy and accuracy ≤ 20% and signal-to-noise ratio > 10:1. LOD is a concentration with signal-to-noise ratio > 3:1.
The stability of plasma MPA was evaluated in QC samples at 0.75, 4, 8 μg/mL, after 12h at RT. The stability of MPA and IS in the autosampler was evaluated after 24h at 4 °C. MPA and IS were stable in plasma if the change in concentration did not exceed ± 15% from the original concentration.
3. RESULTS
MPA extraction from plasma was performed by using SPE. After transferring the samples onto cartridge, SPE was washed once with 1 mL of 5% methanol and then eluted once with 500 μL of pure methanol. MPA signals were detected with a high intensity by HPLC system. This indicated that washing steps were necessary to remove impurities from plasma MPA.
Selectivity: Retention times of MPA and visnadine in standard solutions and QC samples were the same. The spectra of MPA and visnadine in the QC samples were similar to those in the standard solutions. The retention time is 6.456 minutes for MPA and 10.791 minutes for visnadine, respectively. No interference was detected at retention times of interests in blank samples (Figure 1).
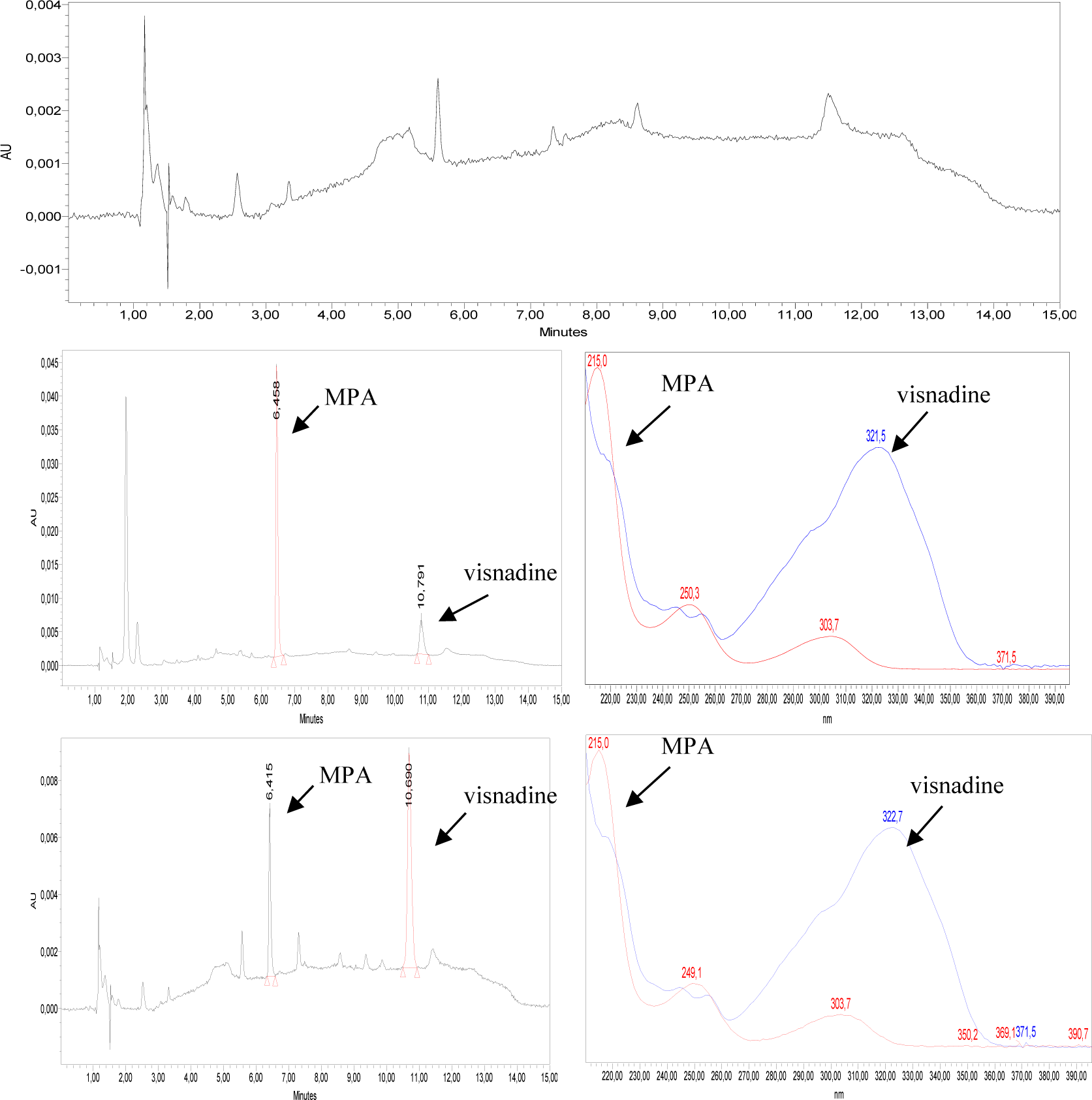
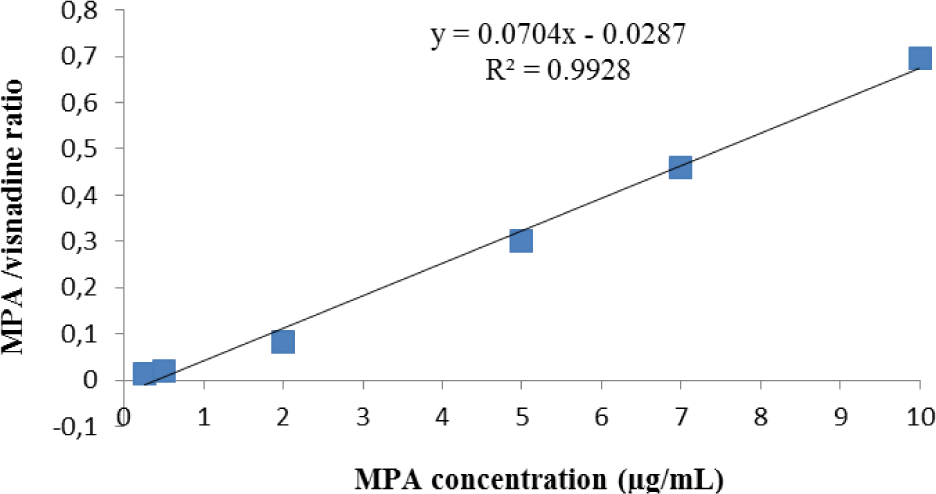
Linearity: The calibration curves of MPA were ranged from 0.25 to 10 μg/mL. The typical linear regression equation between MPA concentration (μg/mL) and ratio of MPA/visnadine peak area was: y = 0.0704x - 0.0287 with the mean value of coefficient of determination (R2) for MPA calibration curve > 0.99 (Figure 2).
Accuracy and Precision: The between-day and within-day accuracies were in the range from 100.04-106.65% at three QC concentrations. The between-day and within-day precision levels were > 92% at all QC concentrations, (CV%, from 2.73% - 7.44%). All inter- and intra-day assays are summarized in Tables 1.
Recovery: Average recoveries at three QC concentrations were > 91% (see Table 2).
Limit tests: LOD and LLOQ concentrations were 0.075 and 0.25 μg/mL, respectively.
Stability: MPA degradation was completely not found after 24 hours in RT (see table 3).
4. DISCUSSION
Mycophenolic acid (MPA) is a widely used immunosuppressant for the purpose of prophylaxis of allograft rejection in transplant patients, nevertheless it has many side effects. Determination of MPA levels in plasma helps to adjust MPA dosing.
Currently, many quantitative methods have been developed to determine MPA plasma concentrations such as EMIT (enzyme multiplied immunoassay technique), HPLC (high-performance liquid chromatography), UPLC (ultra-performance liquid chromatography), LC-MSMS (liquid chromatography–tandem mass spectrometry) [4-7]. Most of chromatographic or immunoassay techniques are used to quantify MPA and its metabolites. The choice of a feasible analytical method depends on biological samples, sample preparation, stationary phase, mobile phase.9 Previous studies showed that the EMIT method is less specific than HPLC due to metabolites cross-reactivity, particularly the acyl glucuronide metabolite of MPA (AcMPAG), and to calibration bias. The EMIT assay displayed therefore slightly higher concentrations than liquid chromatography [10, 11].
Various chromatography methods were developed to simultaneously determine MPA and its metabolites in human plasma [4, 7, 12-15]. Currently, LC-MSMS has been equipped in many large clinical laboratories [15-17]. Although LC-MSMS is considered to be superior to HPLC-UV, it is not devoid of analytical pitfalls. Indeed, the ion suppression effect by interfering components in the matrix could erroneously lead to lower (or higher) results. Furthermore, fragmentation of glucuronide metabolites could lead to falsely results.
Therapeutic drug monitoring of MPA may contribute to prevent acute organ rejection and side effects of drug [3, 5, 17, 18]. Many factors could influence to mycophenolic acid pharmacokinetics such as calcineurin inhibitors and sex [17]. Moreover, a large variability between and within-individual variability resulted in more than a 10-fold variation in the area under the curve of MPA (MPA AUC) [10, 19].
Plasma levels of MPA and more particularly AUCs are correlated with the risk of acute rejection and side effects. Based on HPLC data the proposed target therapeutic window of the total MPA AUC0–12h and predose (C0) concentrations after renal transplantation were defined between 30 – 60 mg.h/L and 1–3.5 μg/mL, respectively [20, 21].
Currently, there are still limitations in applying this method in Viet Nam since no clinical trial is conducted yet (on transplant patients). Therefore, the study is still experimental that needs more clinical confirmation.