
1. INTRODUCTION
Nowadays, with the change of living standards and lifestyle, metabolic disorders such as dyslipidemia (or often hyperlipidemia) and obesity have become serious health problems in the modern world [1]. These diseases can lead to high incidence of morbidity and mortality in both men and women as well as cause an economic burden to the society [1]. Dyslipidemia is characterized by a high levels of total or low-density lipoprotein (LDL) cholesterol, elevated triglycerides and/ or a low levels of high-density lipoprotein (HDL) cholesterol [2]. Dyslipidemia is considered as the biggest contributing factor to the development of athrosclerosis and cardiovascular diseases which is the first cause of death in both developed and developing countries [2]. According to the World Health Organisation, there are about 50% of patients of ischemic heart disease associated with dyslipidemia and more than 4 million deaths every year [3].
Peroxisome Proliferator-Activated Receptors (PPARs) are the nuclear hormone receptors superfamily (class II) [4] including three subtypes, PPARα, PPARβ or δ, and PPARγ, respectively [5]. The PPARs are ligand-activated transcription factors which play a crucial role in the regulation of metabolic processes, energy homeostatis by inducing or repressing target genes [6]. Each subtype displays distinct functions in different cell types [5]. The PPARα was known for controlling and regulating lipid metabolism and inflammation [7]. This receptor is expressed in tissues which have high rate of fatty acid catabolism and in tissues involved in lipid oxidation such as cardiac muscle, liver, kidney, skeletal and adrenal glands [8]. The PPARδ appears in skeletal muscle and adipose tissues which is best known for skin homeostatis [8] and for regulatation of cholesterol, adipogenesis metabolism, and colon cancer [9]. The PPARγ is present in adipose tissue, vascular smooth muscles and immune cells repsonible for energy storage [5] by inducing lipogenesis and fat storage in the tissue as well as improving insulin sensitivity in skeletal muscle [10].
All three PPARs are regulators of numberous metabolic pathways, hence there is a huge increase in the development and use of the PPARs agonists, especially PPARα agonists as main therapeutics of dyslipidemia during the last decade [8]. Number of current marketed drugs for dyslipidemia treatment targets the PPARs. For example, fibrates drugs are activators of PPARα to reduce triacylglycerols used for the treatment of dyslidemia and thiazolidinediones as activatiors of PPARγ for treating hyperglycemia in the type-2 diabetes. Beside, there are other molecular targets for anti-dyslipidemia such as inhibitions of cholesteryl ester transfer protein, cholesterol absorption by binding to cholesterol transporter NPC1L1 (Niemann-pick C1-like1) protein, cholesterol O-acyltransferase enzymes involved in re-esterification of absorbed cholesterol, cholesterol-metabolizing cytochrome P450 or activtion of AMP-activated protein kinase and omega-3 fatty acids, etc [8]. However, there is still rarely drug compounds for those targets [8]. Thus, the PPARs, in particular PPARα has been the main target for anti-dyslipidemia.
The three-dimensional structures of human PPARα in complex with several agonists were solved (PDB codes: 5HYK, 4BCR, 3SP6, 3KDT, 3G8I, 3FEI, 3ET1, 2REW, 2GTK, 1I7G). The structure of human PPARα is very similar to both of the PPARγ and PPARδ [5]. The structure includes four functional domains (A/B, C, D, E/F) (5), in which there are the ligand-independent activation function 1 (AF1) in the A/B domain; the conserved central DNA binding domain in the C domain; docking site for cofactors in the D domain and the ligand binding domain in the E region [5]. The PPARs have multiple binding sites depending on various binding ligands [11]. There are two known binding sites, the main binding pocket for full agonistic and the second one for partial agonistic activities on the PPARα [12].
The ligand binding domain of PPARα is a 12-helix which forms a large hydrophobic pocket [5] (T-shaped cavity) with the volume of about 1300 Å3 [13]. The central pocket spans the region located in between the C-terminal helix 12 (forming one side of a second activation function, AF2 helix) and the 3-stranded antiparallel β sheet [13]. After ligation with the agonist such as fibrate drugs, PPARα undergoes conformation changes [6, 14]. The conserved hydrogen bonds involving the AF2 helix were formed between the protein and the agonists which was found as the most structural differences between the apo protein and the agonist-bound protein [13]. The full agonists stabilise an active conformation of the AF2 helix and enhances heterodimerization process with the retinoid X receptors, promoting recruitment of nuclear receptor coactivators and gene transcription [12, 13]. In addition, a loss of this stabilizating interactions suggested the partial agonist [14]. Without ligands, the PPARα binds to promoters of the target genes leading to transcription repress [6].
However, fibrate drugs as PPARα agonists demonstrated to cause some side effects such as hepatomegaly or liver dysfunction, gastrointestinal disturbance, increase of creatinine levels, etc. [15]. As the results, there is necessary to explore novel compounds for the anti-hyperlipidemic activity with less toxicities. Some medicinal plants with bioactive compounds have been reported for anti-lipid effects as rich source for new effective and safe medicines [16-20] despite of required sufficient evidence for their activities. For example, anthraquinones from Rheum officinale; ginsenoside, ginseng, and polysaccharides from Radix ginseng; triterpenes from Rhizoma alismatis,... reduced triglycerides, LDL-C [19]. Furthermore, some compounds were reported as PPARα agonists such as picrasidine C (an alkaloid from the root of Picrasma quassioides); bixin (a carotenoid from the pericarp of the seeds of Bixa orellana); naringenin (flavanones from the dried, immature fruit of Citrus aurantium); secoiridoid excelside B and some metabolites from Fraxinus excelsior L.etc [20]. Therefore, this study aimed to discover the plant-derived natural compounds against PPARα as PPARα agonists to assist in drug design for anti-dyslipidemia. By using molecular docking for screening, 142 investigated compounds from 29 medicinal plants were docked into the PPARα. The results of ligand binding affinities and their interactions with this receptor were combined to select the potential natural compounds as potential lipid lowering agents.
2. MATERIALS AND METHOD
Molecular docking was applied in in silico screening to select natural compound with high binding affinity into the PPARα. Initially, the diverse bioactive compounds belonging to four main groups, flavonoid, terpenoid, alkaloid and saponin, respectively with their lower-lipid effects were selected for this study. To assist for drug discovery in the next step, the medicinal plants were then searched for containing the compounds. In total, there were 142 natural compounds from 29 medicinal plants including alkaloid (51 compounds), flavonoid (36 compounds), terpenoid (33 compounds) and saponin (22 compounds) were chosen [20-27]. Molecular docking was conducted to investigate binding interactions and to reveal potential natural compounds for treatment of lipid disorders using AutoDock Vina software version 1.1.2 (an open-source program) [28].
Preparation of protein: The 3D crystal structure of the complex PPARα was retrieved from the Protein Data Bank (PDB ID: 5HYK – resolution: 1.83 Å, https://www.rcsb.org) [6]. The structure of PPARα has a co-crystallized ligand-2-methyl-2-[4-(naphthalen-1-yl)phenoxy]propanoic acid (or AL26-29) as the full agonist [6]. The residues making up the main binding pocket are Cys276, Thr279, Ser280, Tyr314, Ile317, Met330, His440 and Tyr464 [6] which covered the co-crystallised-ligand. Using AutoDock Tools 1.5.7rc1, this receptor was prepared for docking; with removing all water molecules and heteroatoms and adding polar hydrogen atoms and Kollman charges into the protein structure. Redocking was carried out by extracting the co-crystallized ligand from the experimental structure and docking the ligand into the binding pocket of PPARα encompassing the native ligand.
Preparation of ligand: Ligands (142 compounds) were prepared in 2D by Chem3D Ultra program (CDX file). Open Babel was employed to convert all 2D structures to 3D structures whose energies were subsequently minimized with the YASARA Energy Minimization server (https://www.yasara.org).
Docking parameters: The docking parameters included the coordinate parameters of the center x, y, z of 7.57 Å; 32.34 Å; 23.882 Å. The grid box was centered on the native ligand with the dimension of 24 x 24 x 24 (Å)3; spacing distance = 1 Å; with default exhaustiveness = 8.
Evaluation of docking results: The docking results were evaluated by the ligand binding affinities (kcal.mol-1), binding pose and the possible interactions between the key residues for biological activities in the target such as Tyr314 for maintaining protein active conformation of protein and for selectivity ligands. In addition, the fibrate drugs as the PPARα agonists including fenofibrate, gemfibrozil, clofibrate, bezafibrate, ciprofibrate were used for docking into the active site of the receptor as reference drugs. The lower binding affinity, the better ligand. BIOVIA Discovery Studio Visualizer 2020 (a free version, downloaded from the website https://discover.3ds.com/) was used for visualisation, assisting in analysis and creating images of 3D models of the protein and the binding mode of protein-ligand.
3. RESULTS
To evaluate the docking protocol, redocking of the co-crystallized ligand into the main binding site of PPARα was conducted. The results showed that this native ligand (the naphthalenic derivatives) bound well into the PPARα with good binding affinity (-11.3 kcal.mol-1) and the root mean square deviation (RMSD) between the docked structure and the native one, using only movable heavy atoms (i.e., only ligand atoms, not hydrogen), was 1.46 Å (less than 2.00 Å). The binding mode and interactions of this ligand with the PPARα were mostly similar to the experimental structure. For example, the polar head of the ligand could form the hydrogen bonds with the side chains of key residues of the active site of the PPARα such as Tyr464 on the H12 helix responsible for agonistic activities towards PPARα and Tyr314, and with the OH of Ser280 [6]. The ligand also interacted with the PPARα through hydrophobic interactions with Phe273 and Phe351. These results demonstrated the reliability of the docking program, so the docking protocol could be then used for screening process.
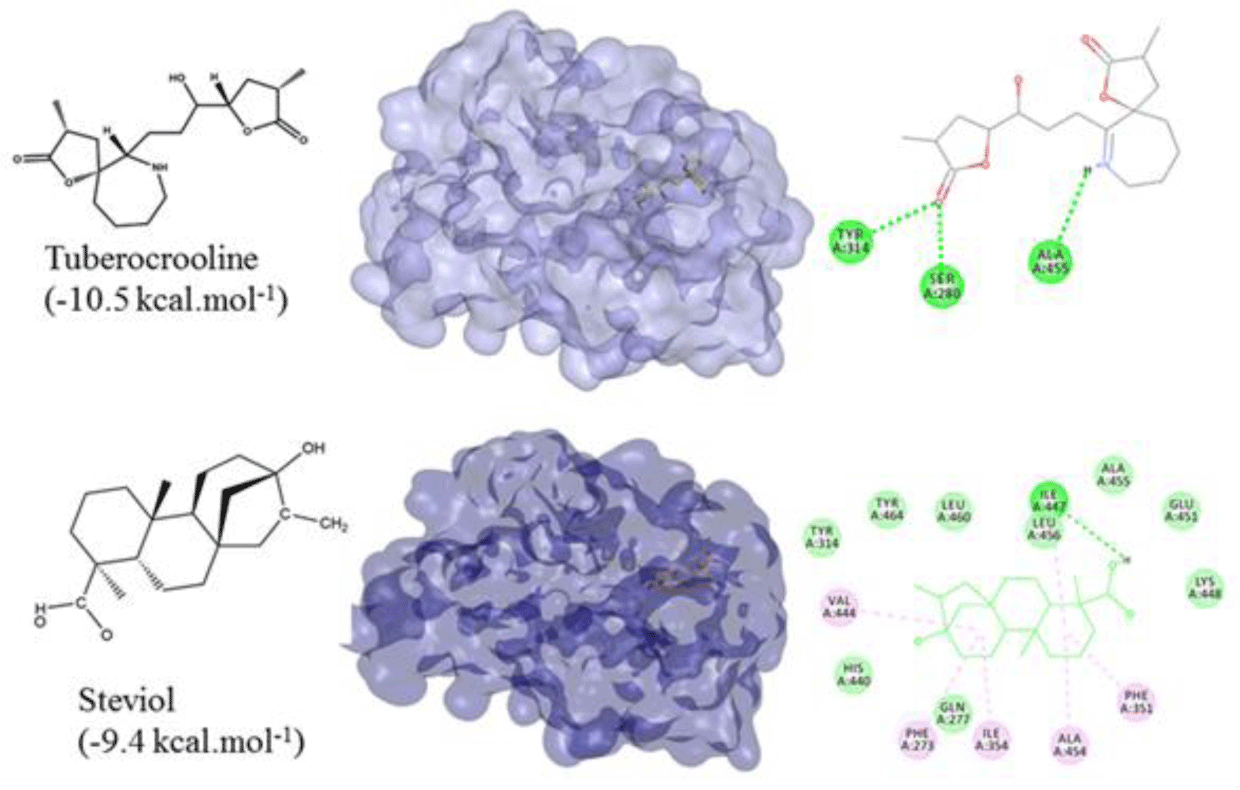
All of 142 natural compounds of the medicinal plants were located into the main binding pocket of PPARα (PDB id: 5HYK) in docking which showed their potential activities against PPARα. These compounds were classified into the subgroups: alkaloid (51 compounds), terpenoid (33 compounds), flavonoid (36 compounds) and saponin (22 compounds) for binding analysis. The results were also compared with those results obtained with the agonisit ligands such as fibrate drugs as reference compounds of PPARα. It showed that flavonoid was the best group of binding well into the PPARα compared to the other groups. Notably, total of 34/142 natural compounds had high affinity for binding into the PPARα (binding affinities of less than -8.0 kcal.mol-1), including 20 flavonoid compounds, 4 terpenoid compounds and 10 alkaloid compounds (Table 1). Of which, the top compounds for each group included: flavonoid group: formononetin from Thermopsis alterniflora (-10.2 kcal.mol-1), diosmetin from Musa spp. (-10.1 kcal.mol-1), luteolin from Elsholtzia ciliate (-9.9 kcal.mol-1); one terpenoid compound: steviol from Stevia rebaudiana (-9.4 kcal.mol-1); and one alkaloid compound: tuberocrooline from Stemona tuberosa (-10.5 kcal.mol-1) (Figure 1 and 2). These compounds strongly accommodated and formed good interactions with the residues of the PPARα binding site as the reference drugs did. Structure and binding affinity relationships of the ligands were also taken for analysis. However, there were four compounds, namely saponarin, vincenin-2, stemonone and tuberostemoenone did not show negative binding affinities as the ligands did not accommodate fully in the binding pocket.

Flavonoid compounds such as formononetin (-10.2 kcal.mol-1) and diosmetin (-10.1 kcal.mol-1) had good binding affinities to the binding pocket of PPARα which showed the potential agonistic activities. This was explained by the presence of -OH groups in the structure of these flavonoids created the hydrogen bonds with four key residues of the receptor such as Ser280, Tyr314, His440 and Tyr464 [6] as well as the aromatic rings made hydrophobic interactions with the target residues which led to the tightly attachment of the compounds into the binding site of PPARα. These results were compatible with the experimental results that PPAR agonists interact to form similar interactions with polar residues, especially with Tyr464 localized in AF2 which was important for stabilizing the active conformation of protein [6]. In addition, the three other flavonoids such as luteolin (-9.9 kcal.mol-1), daidzein (-9.7 kcal.mol-1), and chrysin (-9.6 kcal.mol-1) were also good ones towards the binding affinities on the PPARα. The ligands with the simple backbone could attach the hydrophobic pocket better than the isoflavon and flavon compounds with the presence of glucosides or alkyl groups, for example: luteolin (-9.9 kcal.mol-1) > luteolin-7-rutinoside (-7.2 kcal.mol-1); daidzein (-9.7 kcal.mol-1) > daidzin (-9.1 kcal.mol-1); and apigenin (-9.4 kcal.mol-1) > apigenin-7-glucoside (-8.9 kcal.mol-1). The results could be related to the experimental resuls that the aglycone penetrates easily due to the high lipophilicity and low molecular weight [29]. The other isoflavon and flavon compounds with the presence of glucoside or alkyl groups such as daidzin, thermopsoside, ononin, isoangustone, apigenin-7-glucoside, saponarin 4-O-glucoside, licoflavonol, kumatakenin did not show strong binding affinities (Table 1). Structures of the eight compounds with the good binding affinities of less than - 8.0 kcal.mol-1 and their binding modes into the PPARα (PDB id: 5HYK) were presented in Figure 3 and 4.


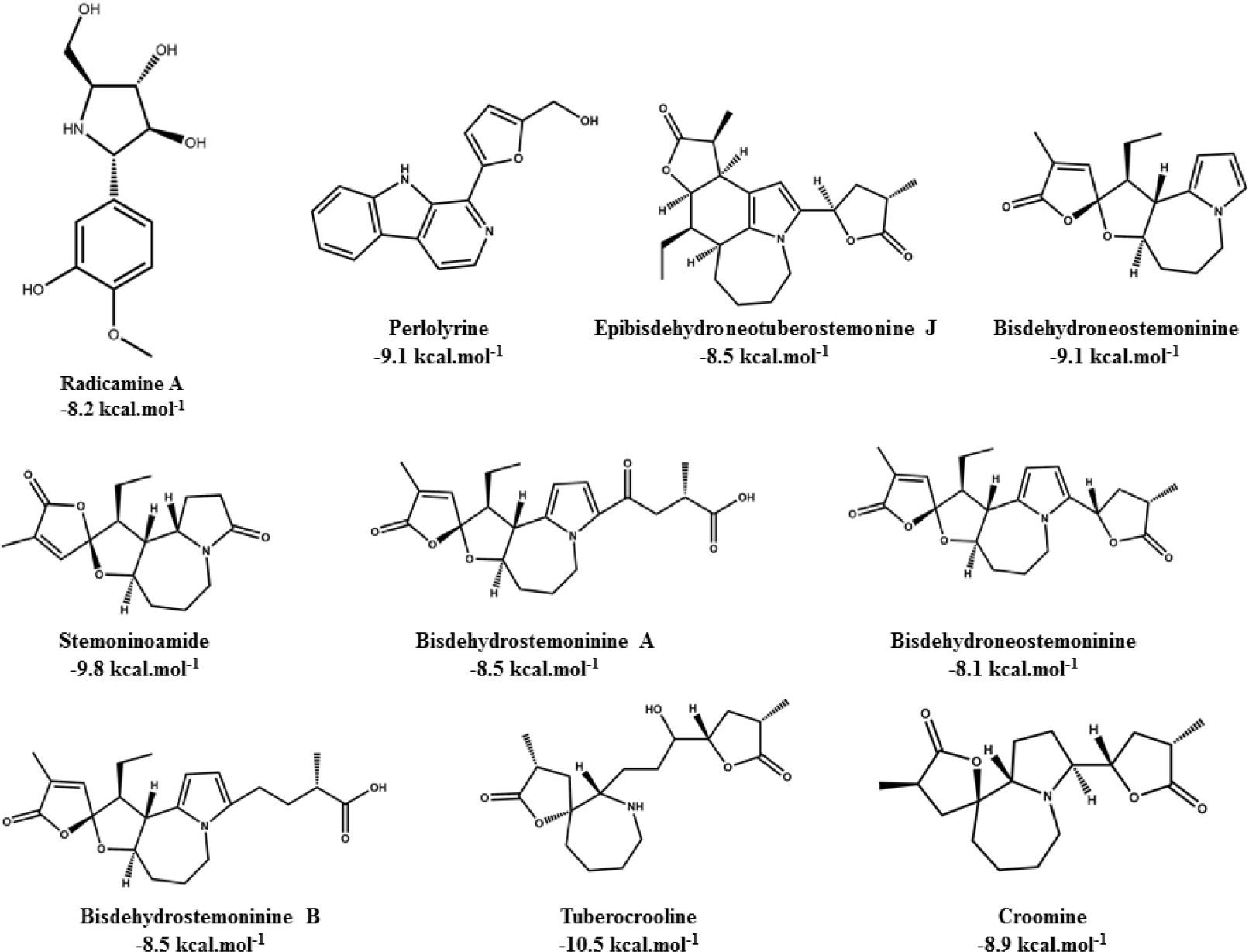
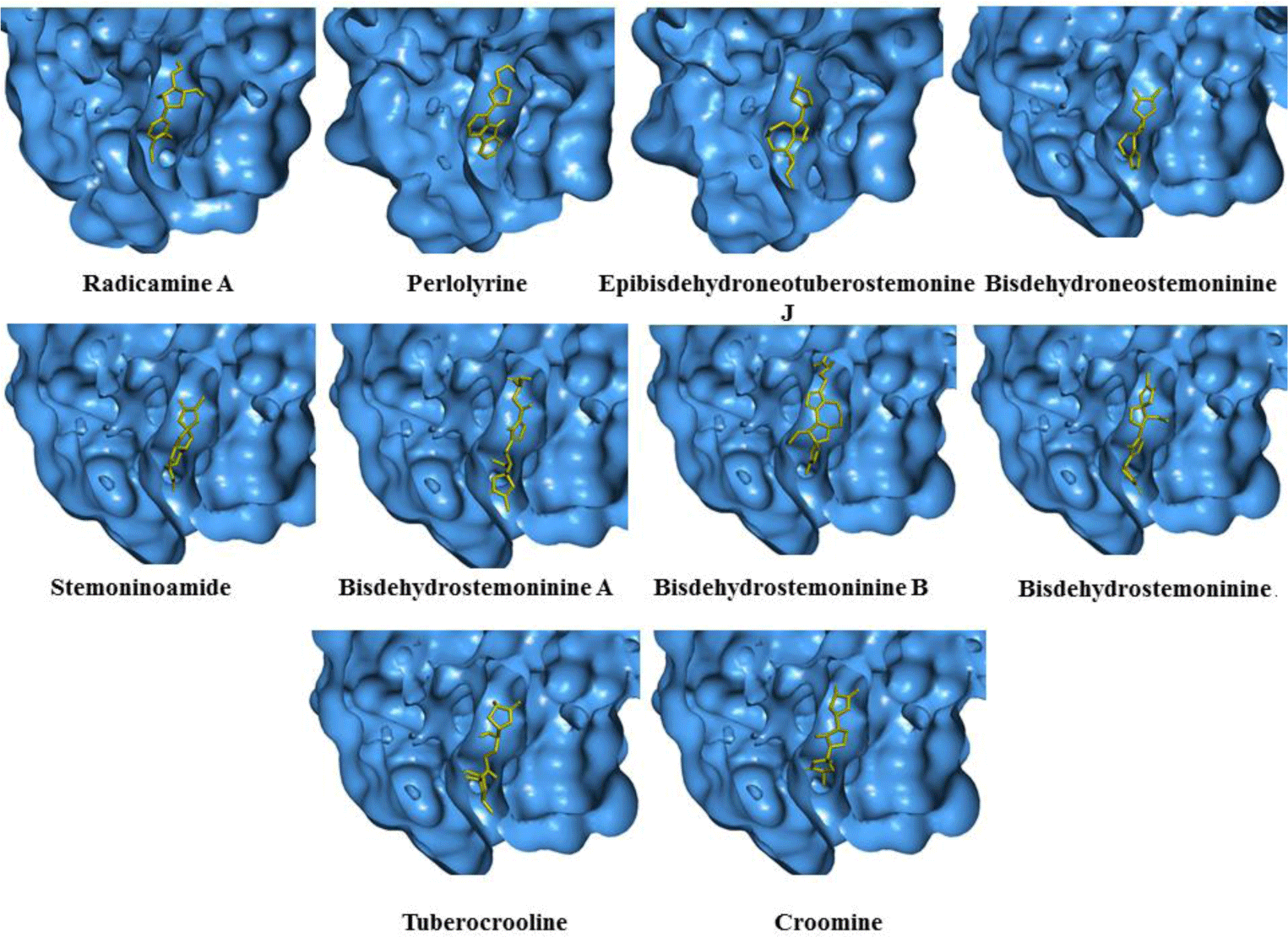
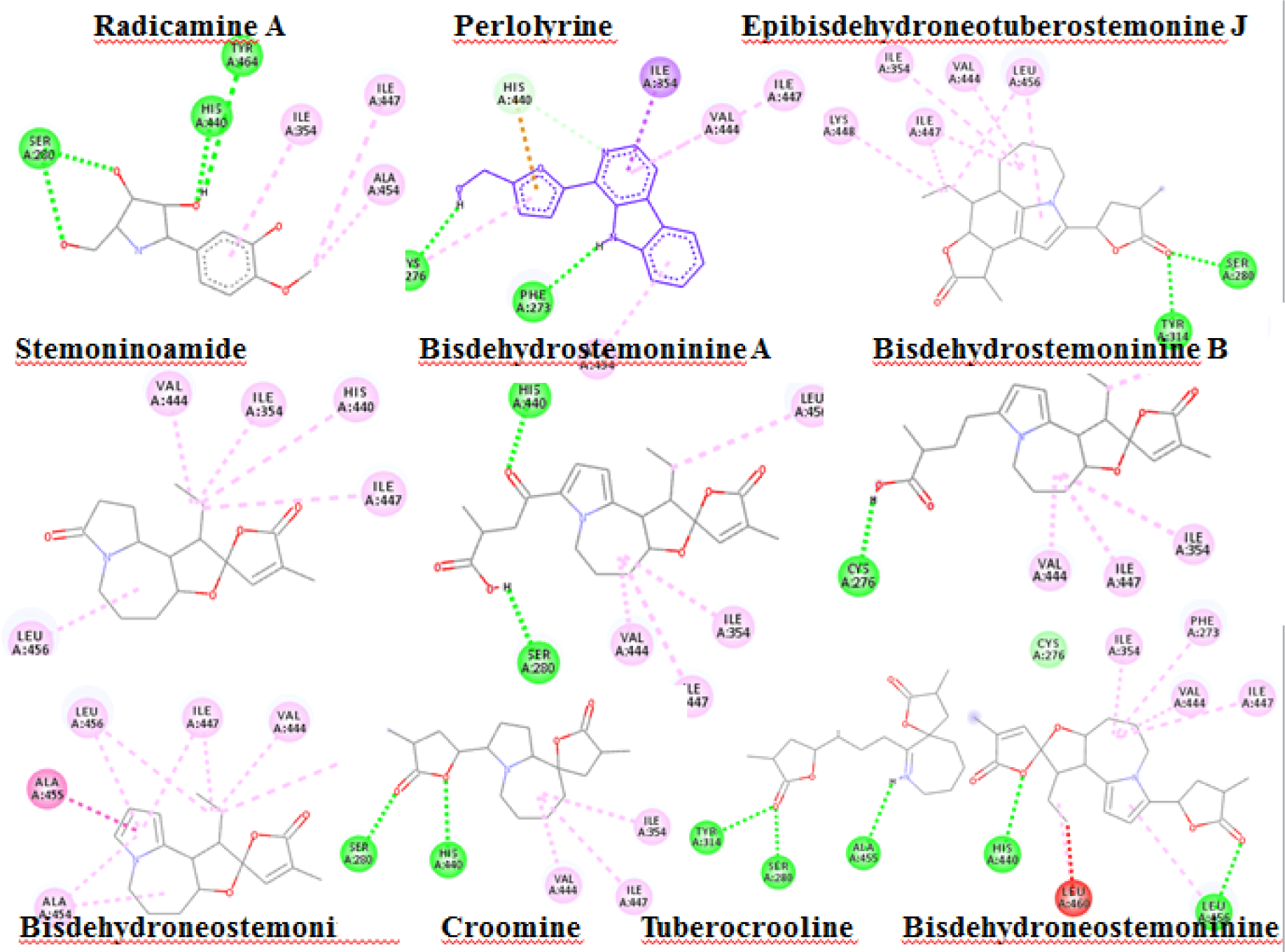
Alkaloid compounds bound to the PPARα with binding affinities ranging from -1.0 to -10.5 kcal.mol-1. Ten alkaloid compounds including tuberocrooline (-10.5 kcal.mol-1 had good affinities on the PPARα due to forming the hydrophobic interactions between the aromactic rings containing nitrogen and some important amino acids of binding site such as His440 (for example, perlolyrine: -9.1 kcal.mol-1). The compounds also generated the hydrogen bonds through their side chains having some functional groups (OH or COOH groups) with residues Ser280, Tyr314 and His440 of PPARα (for example, radicamine A: -8.2 kcal.mol-1). These interactions were shared by all PPARα agonist like fibrates and the other ligands reported in the PDB [6, 12], with data showed in Table 2. The alkaloid possess the simple aromactic ring like radicamine A and perlolysine could attach further in the binding pocket than the ligands having bulky aromatic groups. However, replacing pyrrole ring into pyrrolidine ring made the compound not going deeper into the cavity or extending the length of side chain of pyrrole which led to reduce the binding affinities, such as the case of compound 9a-bisdehydrotuberostemonie with the pyrrole ring (-7.0 kcal.mol-1) > neotuberostemonol with the pyrrolidine ring (-4.2 kcal.mol-1), and stemoninoamide (-9.8 kcal.mol-1) > bisdehydrostemonine A and B (-8.5 kcal.mol-1). When pyrrolidine opens, for example tuberocrooline (-10.5 kcal.mol-1), the compound made the C=O group of tetrahydrofuran interacted with Tyr314 and also formed more hydrogen bond with the residue Ala455 of target, so this compound showed strongly affinity than, croomine (-8.9 kcal.mol-1). All of Figure 5, 6 and 7 illustrated 10 top alkaloid compounds, namely radicamine A, perlolyrine, epibisdehydroneotuberostemonine J, bisdehydroneostemoninine, stemoninoamide, bisdehydrostemoninine A, bisdehydrostemoninine B, bisdehydrostemoninine, tuberocrooline, croomine, respectively with their good binding affinities of less than -8.0 kcal.mol-1 and their binding modes into the PPARα.
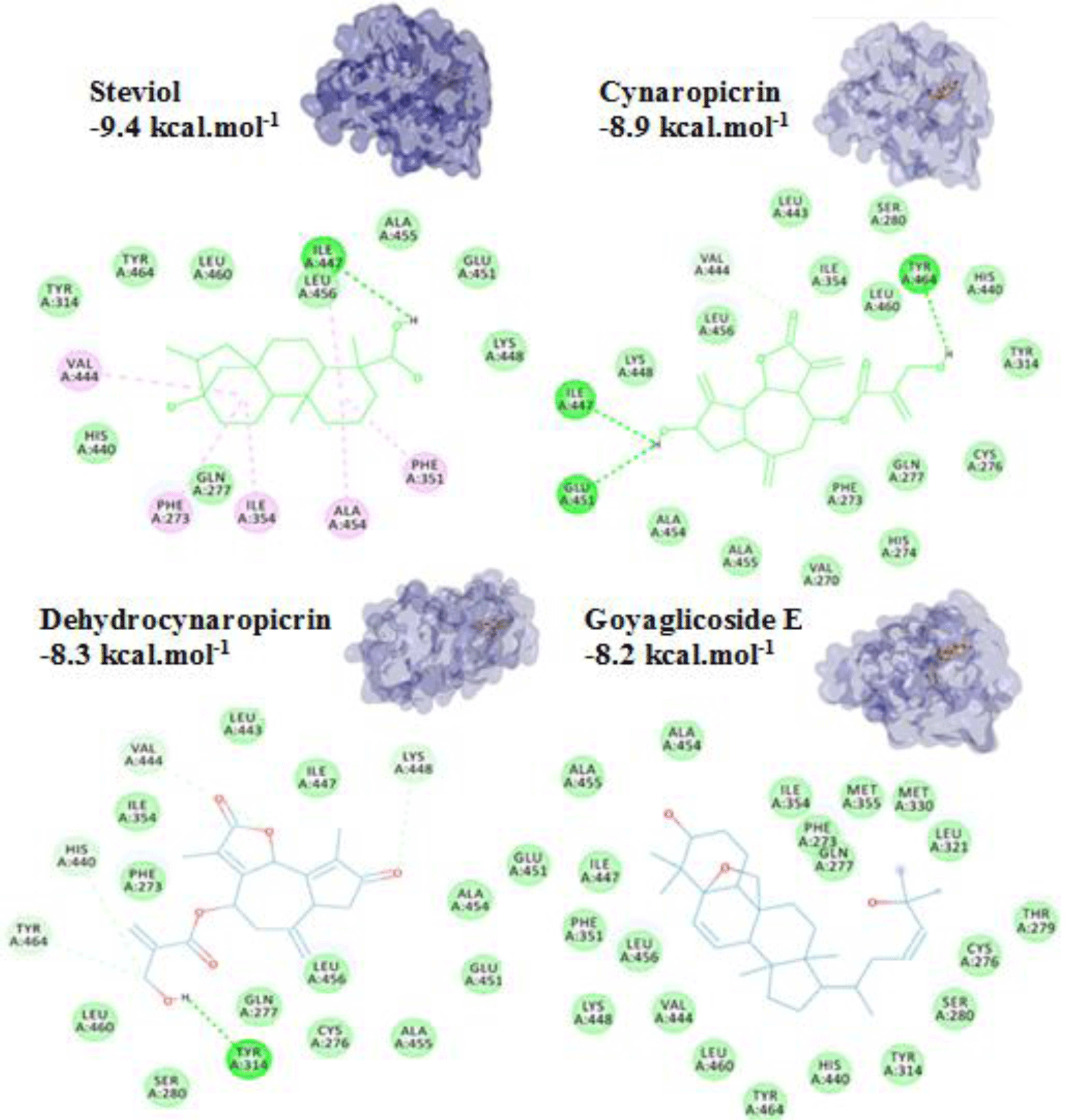
For terpenoids and alkaloids, the compounds belonging to these groups could get into the active site of the PPARα, however, their binding affinities were not as good as the other groups. Of which, three terpenoid compounds, namely steviol, cynarpicrine and dehydrocynaropicrine and goyaglycoside E interacted well with the residues Tyr314, Tyr464, Ile447 at the active site of the PPARα by hydrogen bonding with their affinities of -9.4; -8.9, -8.3 and -8.2 kcal.mol-1, respectively (presented in Figure 8). It could be seen that the presence of OH group in the structures such as steviol, cyanopicrin, dehydrocyanopicrin increased the binding affinities. The results agreed with the experimental data that -OH group was essential for the activities in terpenoids (20). Only one saponin compound, diosgenin displayed good binding affinity of -7.5 kcal.mol-1. The saponin ginsenosides, such as ginsenoside Rb1 did not show high binding affinity (-5.5 kcal.mol-1). The experimental results also reported that the compound responsible for the bioactivities of ginseng which demonstrated the inhibition of PPARα in vitro and in vivo with a consequent increased serum concentrations of total cholesterol, triglycerides, and HDL cholesterol [30].
Moreover, the docking results of fibrate drugs as the PPARα agonists (fenofibrate, gemfibrozil, clofibrate, bezafibrate, ciprofibrate) into the PPARα as reference drugs showed that these drugs bound closely to the binding site of the receptor with high binding affinities: fenofibrate (-10.2 kcal.mol-1), gemfibrozil (-8.2 kcal.mol-1), clofibrate (-7.3 kcal.mol-1), bezafibrate (-9.1 kcal.mol-1), ciprofibrate (-8.6 kcal.mol-1) (Table 2). Comparison of docking results of the 142 natural compounds and the reference drugs, it showed that most of the investigated compounds bound to the main binding pocket and strongly mimicking the observed interactions like the reference ones did. The reference drugs interacted well with the receptor by hydrogen bonds and hydrophobic interactions with some important residues such as Tyr464 important for recruiting co-activators (6) and Phe273, Tyr314, Phe351 and Tyr464. In particular, the standard polar interactions with Ser280, Tyr314 and Tyr464 presented in the all agonists complexed to the receptor (6) which were found in all of the top investigated compounds.
Overall, top compounds selected from each group including formononetin, diosmetin, luteolin, daidzein, chrysin (flavonoid compounds); steviol (terpenoid compound) and tuberrocrooline (alkaloid compound) possess high binding affinities. They occupied the same position and shared ability to form interactions with the residues of binding pocket of the PPARα. However, taking into account of forming hydrogen bonds network with the key residues Tyr464, His440, Tyr314 and Ser280 for agonistic activities, the following compounds, namely formononetin from Thermopsis alterniflora (-10.2 kcal.mol-1), diosmetin from Musa spp. (-10.1 kcal.mol-1), luteolin from Elsholtzia ciliate (-9.9 kcal.mol-1); steviol from Stevia rebaudiana (-9.4 kcal.mol-1); and tuberocrooline from Stemona tuberosa (-10.5 kcal.mol-1) were selected. They could be used as the potential PPARα agonists for further biological testings for their anti-dyslipidemia effects.
4. DISCUSSION
The natural compounds from medicinal plants have been of interest in drug discovery for the treatment of many diseases, including dyslipidemia in recent years. Therefore, in this study, structure-based virtual screening through molecular docking was conducted for 142 investigated natural compounds into the PPARα (PDB id: 5HYK) to discover potential compounds for anti-dyslipidemia. Through the criteria including ligand binding affinities, binding modes and binding interactions, flavonoid compounds performed the most potential compounds. These compounds formed favorable bindings with the PPARα by hydrogen bonds with key residues of the target, Ser280, Tyr314 and Tyr464, crucial for agonistic activities. Of which, two flavonoids, formononetin from Thermopsis alterniflora (-10.2 kcal.mol-1), diosmetin from Musa spp. (-10.1 kcal.mol-1) and luteolin from Elsholtzia ciliate (-9.9 kcal.mol-1) were the top compounds of high binding affinities which suggesting they might be potential compounds as PPARα agonists for further testing toward anti-dyslipidemia effects.
This results were consistent with the reported results from the experiments that formononetin was the most potent activators of PPARα with the value of EC50 of less than 1.0 mol/L [20, 31]. Diosmetin demonstrated inhibition of fat accumulation in liver and epididymal tissues, however diosmetin had little effect on the parameters related to dyslipidemia [32]. Diosmetin (3’,5,7-trihydroxy-4’-methoxy-flavone), a flavone aglycone have been displayed to have several pharmacological effects such as anti-inflammatory and hepatoprotective role in an in vivo model [33] or antihyperglycemic activities [34]. Another study reported that diosmetin treatment inhibited fat accumulation in liver and epididymal tissues and improved glucose intolerance by lowering glucose levels during a glucose tolerance test [32]. Furthermore, luteolin has been shown to have anti-inflammatory activity as well as to protect against tumor necrosis factor-alpha and especially to prevent lipid accumulation and decrease total cholesterol, triglyceride and LDL-cholesterol levels [35]. Therefore, the findings in this study suggested the mechanism of these compounds toward to PPARα.
Moreover, two other compounds, one terpenoid compound: steviol from Stevia rebaudiana (-9.4 kcal.mol-1); and one alkaloid compound: tuberocrooline from Stemona tuberosa (-10.5 kcal.mol-1) were also identified as the hit natural compounds for PPARα agonists. Steviol, the terpene aglycone, the final metabolite of all steviol glycosides was reported with some bioactivites such as antidiabetic effects on streptozotocin-induced diabetic mice [36], antihyperglycemic, antihypertensive, antioxidant and anti-inflammatory effects, etc [37]. Steviol causes a decrease in glucose accumulation in intestinal ring tissue, liver and kidney and also enhances insulin secretion [36]. Tuberocrooline has not been demonstrated for anti-dyslipidemia, but this compound was known for the antitussive activity [38]. Furthermore, the formononetin has been proved for anticancer activity [39], diosmetin for the biological activity on alloxan diabetic rats [40], steviol for antioxidant capacity [41] and tuberocrooline for the acetylcholinesterase inhibitory activity [42].
Therefore, the good compounds were selected, namely formononetin from Thermopsis alterniflora, diosmetin from Musa spp., luteolin from Elsholtzia ciliate; steviol from Stevia rebaudiana; and tuberocrooline from Stemona tuberosa, respectively for further steps in drug discovery and design of PPARα agonists for anti-hyperlipidemia activities. Combination of the experimental results of these compounds for dyslipidemia effects and the results obtained in this study, the current research also suggested the mechanism of these compounds towards to the PPARα for agonistic activities. However, there were still some limits in this study. It is required more number of investigated compounds and partial agonistic on the PPARα or dual agonist/ pan-agonistic activities combining on PPARγ could be investigated in the screening process of better anti-dyslipidemia and also for PPARα agonists compounds.
Conclusion
Metabolic disorders such as dyslipidemia have been in the list of serious health problems in the modern world. This disease is considered as the major contributing factor to the mortality of men and women in both developed and developing countries. With the aims of identifying the potential plant-derived natural compounds as PPARα agonists for assissiting in drug discovery of anti-hyperlipidemia, 142 compounds were docked into the structure of PPARα (PDB ID: 5HYK). The screening results discovered 34 compounds including 20 flavonoid, 4 terpenoid and 10 alkaloid compounds strongly accommodated and formed good interactions with the residues of the PPARγ binding site as the reference drugs did. Flavonoid was the best compounds attaching into the binding site of the PPARα. Top compounds for anti-dyslipidemia were identified, namely formononetin from Thermopsis alterniflora, diosmetin from Musa spp., luteolin from Elsholtzia ciliate; steviol from Stevia rebaudiana; and tuberocrooline from Stemona tuberosa. These compounds formed favorable interactions with the binding site of PPARα through hydrogen bonds and hydrophobic interactions with key residues of the target, Ser280, Tyr314 and Tyr464, leading to agonistic activities. Combination of these results and the experimental results of these compounds for dyslipidemia effects, the current study also suggested the mechanism of the compounds as the PPARα agonists. Therefore, this study provided useful information for further drug discovery and design of potential lipid-lowering agents as the replacement for the current PPARα agonists like fibrate drugs.