
1. INTRODUCTION
Along with the rapid emergence of antibiotic resistance, currently available therapeutic agents are becoming less effective in treating bacterial infections, especially in cases of multidrug-resistant (MDR). Annually, nosocomial infections cause more than 63,000 deaths in the United States of America, whereas MDR bacteria are responsible for approximately 25,000 deaths in Europe [1]. This intense battle against MDR bacteria necessitates the search for novel antibiotics. Conventional approaches are seen to be unsatisfactory, therefore, innovative strategies are currently being devised to confront the challenges of antibacterial discovery [2]. With recent advances in chemoinformatics and the availability of structural and biological data of macromolecules, computational research is helping in developing new agents that target essential bacterial proteins. The in silico results, in concert with physicochemical and biological experiments, could elucidate the mechanism of drug action as well as the mechanism of drug resistance of bacteria, facilitating the process of drug discovery [3].
In the last few decades, 4,5-dihydro-1H-pyrazole derivatives, commonly known as 2-pyrazoline, have agents thanks to their wide range of biological activities. 2-Pyrazoline derivatives exhibit various remarkable activities, including antibacterial, anticancer, anti-inflammatory, antioxidant, antidepressant, etc. [4] Despite numerous explorations on 2-pyrazoline scaffolds, their detailed mechanism of action remains uncleared [5]. Some studies suggested that the antibacterial activity of 2-pyrazoline derivatives is attributed to their ability to inhibit FabH enzyme [6,7]. Furthermore, molecular docking studies have shown good binding affinity of 2-pyrazoline derivatives to this target [8,9]. Recent research has also revealed the great potential of 2-pyrazoline derivatives in developing antineoplastic agents [10-12]. Notably, Hai-Liang Zhu et al. (2015) synthesized 2-pyrazoline derivatives that have IC50 values on BRAF, WM266.4, and MCF-7 (0.05, 0.12, and 0.16 μM, respectively) comparable with sorafenib, an FDA-approved drug for the treatment of kidney, liver, and thyroid cancer (0.03, 0.06, and 0.19 μM, respectively) [13].
β-Ketoacyl-acyl carrier protein synthase III (FabH) initiates the fatty acid biosynthesis by catalyzing the first step between acetyl-CoA and malonyl-ACP to produce β-ketoacyl-ACP [14]. While bacteria utilize monofunctional enzymes for different steps of the fatty acid biosynthesis, the whole process is catalyzed by a large multifunctional protein in humans. This fundamental difference in structure and organization of fatty acid biosynthesis systems in bacteria and in humans has distinguished FabH as a potential target for the development of novel antibacterial agents with minimal side effects in human hosts [15].
In our previous study, we synthesized some 2-pyrazoline derivatives, which showed good activity against S. aureus (MIC values ranging from 4 to 128 μg/mL) [16]. In this study, to predict the structure of high active 2-pyrazoline derivatives, a molecular docking model on FabH protein was built using computational tools and a training dataset of compounds taken from our previous research. This model was then validated using a validation dataset of substances obtained from other studies on 2-pyrazoline derivatives. Compounds with potentially high affinity towards FabH enzyme predicted by the docking were then synthesized and tested for antibacterial activity to evaluate the model’s applicability. In the drug discovery, the screening of large chemical libraries with high structural diversity exponentially increases the probability of identifying initial lead compounds [17]. For this reason, we conducted the cytotoxicity assay in three cancer cell lines, including HepG2 (human liver cancer cells), RD (human rhabdomyosarcoma cells), and MDA-MB-231 (human breast cancer cells), to determine the anticancer activity of 2-pyrazoline derivatives collected from this study and our previous study. This initial screening enriches the anticancer activity database of 2-pyrazoline derivatives, facilitating future studies on this class of compounds.
2. MATERIALS AND METHOD
The crystal structures of protein FabH of E. coli and S. aureus were downloaded from Protein Data Bank [18]. A docking model would be easily built and would be more precise if the target protein has a high-resolution exhibit highly inhibitory activity against the enzyme. Accordingly, we selected the structure 1MSZ as the E. coli FabH (ecFabH), which has a co-crystalized ligand [19], and 1ZOW as the S. aureus FabH (saFabH), thanks to its high resolution at 2.0 Å [20]. The protein structure was first treated by the LigX tool in MOE 2008.10 [21] to achieve the desired conformation, similar to its normal native state.
The training dataset, including fifteen 2-pyrazoline derivatives from our previous study, was drawn and rendered to its lowest energy conformation using Sybyl-X 2.0 [22]. From this library of ligand conformations and the refined protein structure, the docking model was built using FlexX docking tool in leadIT 2.0.2 software [23].
The docking model was created by identifying the position and radius of the binding site. Binding mode was shown by interactions between ligand and amino acids in the binding region. Binding ability was assessed by docking score with the core principle: the more negative the score, the stronger affinity. A rational binding model would correlate the docking scores of ligand test sets with experimentally determined binding affinities. This model could be used to explain the structure-activity relationship and preliminarily predict the biological activity of other scaffold-shared compounds.
Thirty-seven pyrazoline derivatives from three studies were served as the validation dataset of the recently built docking model. We deliberately selected studies that conducted the antibacterial assay on the same bacterial strain in our research, ATCC 25923, to be more precise [24-26]. The molecules from these studies were applied to our docking model, then the docking score would be recorded. This docking score and the logarithm of their experimental MIC value were analyzed using GraphPad Prism 9.1.0 [27], the linear regression was built with the significant level α = 0.05, and the goodness of fit was measured by R-square value.
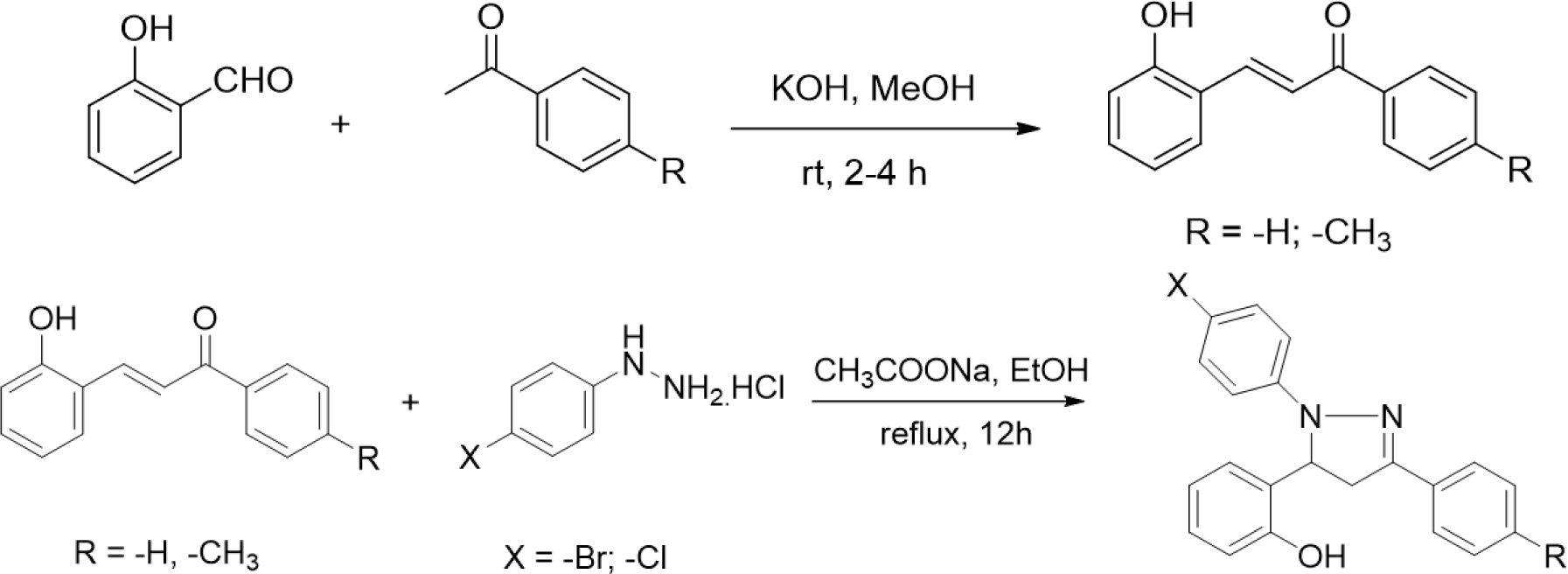
The synthesis of 2-pyrazoline derivatives follows two steps. The first step was the condensation between salicylaldehyde and acetophenone derivatives under strongly basic conditions, resulting in chalcone derivatives. The 2-pyrazoline derivatives were subsequently synthesized by condensation between the purified chalcones and phenylhydrazine derivatives. These two reactions were performed following the procedure described in our previous study [16].
All reactions were monitored using thin-layer chromatography, the products were later purified by recrystallization. The structures of the isolated compounds were confirmed by mass spectrum (MS) and nuclear magnetic resonance spectrum (NMR).
In vitro antibacterial activities against E. coli ATCC 25922 and S. aureus ATCC 25923 were evaluated for all synthesized compounds by using the agar dilution method. The solutions of test compounds were incorporated into a nutrient agar medium to produce the plates in which the final concentrations of the test compound range from 0.5 to 128 μg/mL with a 2-fold difference in concentration between the two plates. A bacterial suspension was afterward dropwised on each plate. The bacterial growth inhibition was assessed after 24h, and the lowest concentration at which no bacterial growth was detected was the MIC value.
Cytotoxicity of selected 2-pyrazolines derivatives was assessed on three malignant cell lines (HepG2, RD, and MDA-MB-231) using the MTT assay. After incubating the cells with a series of concentrations (from 2.5 to 100 μM), the percentage of viable cells was determined through the activity of the enzyme succinate dehydrogenase (SDH) present in intact mitochondria of living cells. In fact, SDH converts MTT [3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide)] to formazan, which is soluble in acidified isopropanol to form a purple solution. The optical density (OD) value at 570 nm reflected the number of viable cells in the sample that was used to calculate the inhibitory percentage in the following equation:
3. RESULTS
FlexX software shows the active site of ecFabH enzyme, including Cys112, His244, and Asn278 (Cys112, His238, Asn268 of saFabH), in the middle of a tunnel with two outlet-like chambers leading out to the surface of the enzyme.
Chamber 1: surrounded by Typ32, Arg151, Ile155, Asn210, Phe213, Arg249. This region is where acyl-CoA attaches to bring the acyl group into the catalytic site [8].
Chamber 2: surrounded by Phe87, Leu142, Leu189, Asn193, Gly307. The width of the chamber determines the size of the acyl group in acyl-CoA substrates. This chamber of saFabH is larger than the one of ecFabH [28].
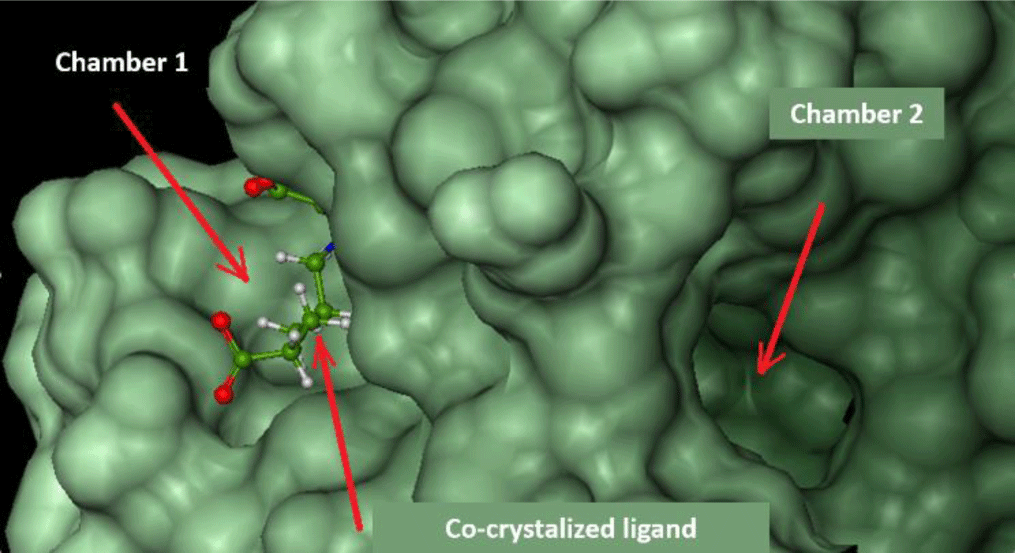
The docking study was conducted on both chambers. Parameters of docking models are presented in Table 2, and respective docking scores are presented in Table 3.
Model 4 shows a high correlation between the docking score and the logarithm of the MIC value of the training compounds (Figure 4). In model 4, hydrogen bonds were formed between hydroxyl, nitrogen atoms of testing molecules with amino acid residues in the binding site. In addition, the N-phenyl substituent of the pyrazoline ring forms hydrophobic interactions with residues in the binding chamber (Figure 5).
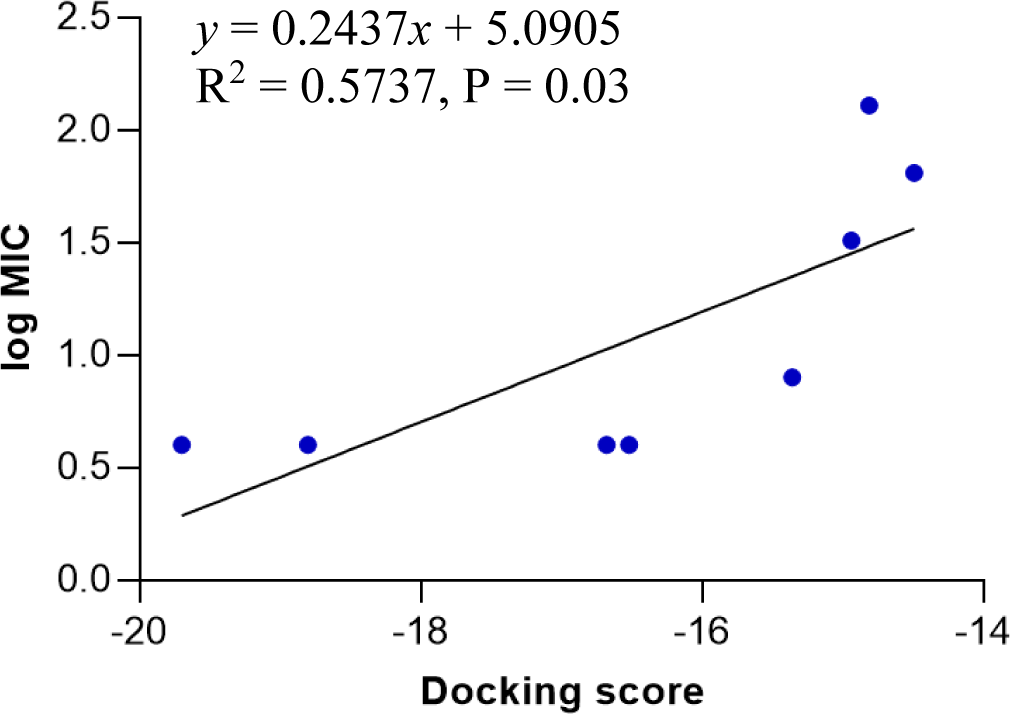

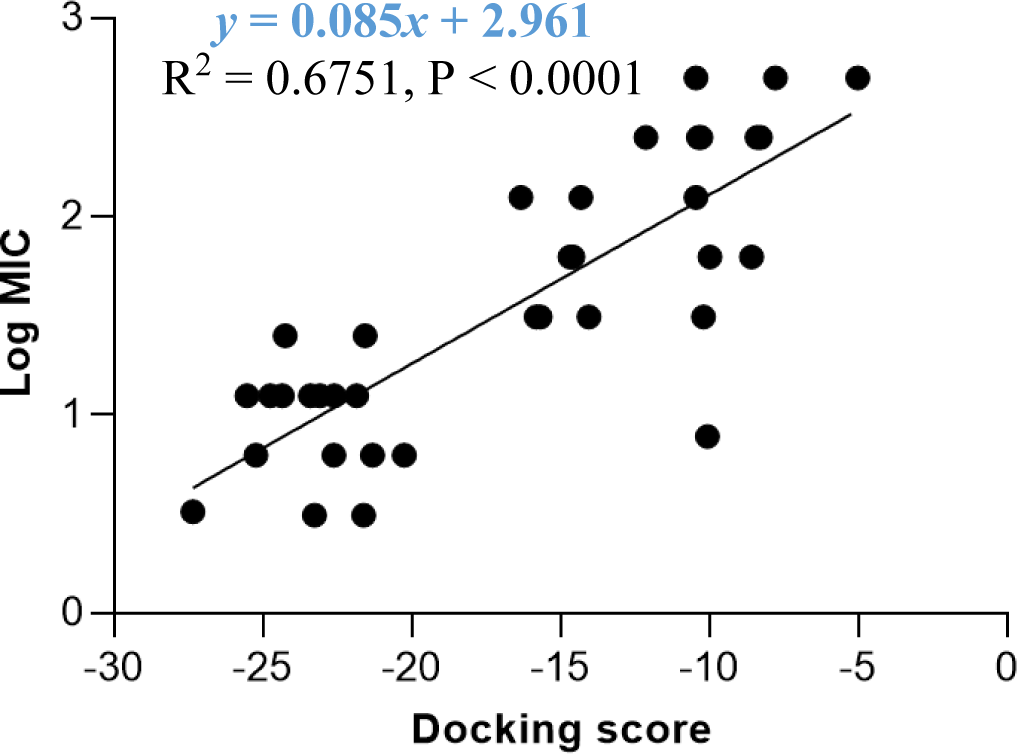
The docking model 4 was validated using a series of structurally diverse 2-pyrazoline derivatives obtained from three different studies as the validation dataset. A strong correlation was observed between the docking score and the log MIC value of these compounds with an R-square value of 0.6751 (p < 0.0001) (Figure 6).
According to the docking model, four 2-pyrazoline-based molecules with docking scores lower than -16 kJ were synthesized and evaluated for antimicrobial activities against E. coli (ATCC 25922) and S. aureus (ATCC 25923). As expected, all compounds do not affect the growth of E. coli and have high activity against S. aureus (MIC 4-16 μg/mL) (Table 3).
In addition, these four compounds, together with fifteen compounds collected from our previous research (the training dataset), were evaluated for cytotoxic activity [16]. All testing compounds exhibit a moderate effect on the three cancer cell lines. Among them, HP10 simultaneously expresses excellent activity on HepG2, RD, and MBA cell lines with IC50 values are 26.62, 17.74, 14.47, respectively (Table 6).
4. DISCUSSION
Among two FabH protein structures downloaded from PDB, only the ecFabH has a co-crystallized ligand. Although the docking model on ecFabH shows that the ligand has a high docking score (-29.44 kJ/mol), this compound shows only moderate activity against ecFabH with an IC50 value of 7 μM [29]. This contradiction implies that the docking model associated with the enzyme active site may not correlate with the inhibitory activity against this enzyme. The binding model associated with chamber 2 (model 4) has the docking score that highly correlates with the log MIC value on S. aureus of the training compounds. The main substrates of saFabH enzyme contain voluminous side chains such as isobutyryl, caproyl, butyryl, isovaleryl-CoA; therefore, binding of ligands on chamber 2 narrows the acyl-harboring space. This narrowed space prevents binding between saFabH enzyme and their substrates. In reverse, ecFabH enzyme primarily binds to substrates with smaller acyl chains such as acetyl-CoA and propionyl-CoA; therefore, the ligand binding at chamber 2 does not affect the enzyme activity, which explains why there was no testing substance active on E. coli [28].
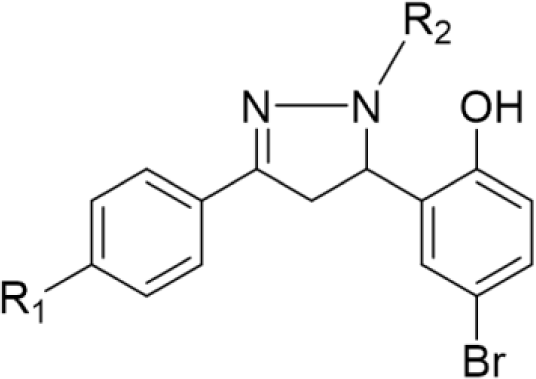
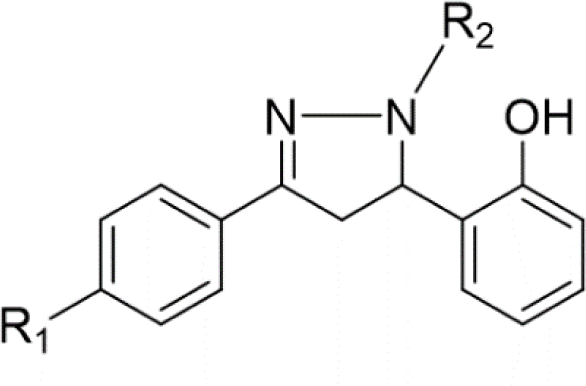
In concern with the structure-activity relationship of 2-pyrazoline derivatives, the binding mode shows that the -OH group on the aromatic ring at C5 of the 2-pyrazoline ring creates hydrogen bonds with amino acids in the active site. This substituent is, therefore, essential for the activity. In addition, the presence of -Br substituent at the para position with the -OH group has a minor effect on the activity, moving this bromo group to N1 of the phenyl ring converted HP6 and HP8 to HP18 and HP19, respectively, in which MIC values drop from 4 to 8 μg/mL. This observation is consistent with the docking model: only the aromatic ring at the N1 position enters a deeper space of the acyl-harboring region (Figure 5).
The docking model 4 was validated with external data. Besides confirming the model’s accuracy, an external dataset with a diversity of substituents on the pyrazoline ring rules out the possibility that the antibacterial activity was caused by other random structural factors. This model was also endorsed by the MIC value on S. aureus of newly synthesized 2-pyrazoline derivatives in this study. In particular, substances with lower docking scores generally show better antibacterial activity than high-docking score compounds. Besides, in vitro MIC values are 2-3 times smaller than in silico MIC values predicted by the docking model. This difference was deliberately acceptable due to the concentration limit upon assay preparation. The high correlation between docking score and antibacterial activity highlights the applicability/reliability of the docking model in predicting antimicrobial activities of other 2-pyrazoline compounds.
Upon analyzing the cytotoxic activity of the nineteen 2-pyrazoline derivatives, the result reveals a similar trend on different cancer cell lines. In other words, when comparing the anticancer activities of two random substances, the one which is considered as more active mostly has better IC50 values in all three cell lines. This reality implies the existence of a common functional enzyme in all three cell lines that was targeted by these 2-pyrazoline derivatives. Compound series HP1-HP5 and HP11-HP15, with hydrogen and acetyl substituents, respectively, present at the N1 position, generally exhibit lower activity compared with substances HP6-HP10 and HP16-HP19, bearing the phenyl group at the N1 position. The N1-phenyl substituent appears to contribute to the overall activity of these derivatives. On examining the activities of the two series HP6-HP10 and HP16-HP19, the slight difference in activity implies that a halogen substituent at the p-position of the aromatic nucleus at N1 has little effect on the activity. The p-bromo group of the phenyl ring at C5 appears to act in the same manner. Changing its position on the aromatic ring or adding hydrophilic groups may influence the activity of these derivatives.
Standing out from the empirical analysis of the structure-activity relationship, our docking model on saFabH provides additional quantifiable insights into the role of different distinctive groups existing in the structure of 2-pyrazoline derivatives toward the whole antibacterial activity of these molecules. The binding mode and docking scores may confer a stronger theoretical basis on the structure modification. However, a robust docking score does not secure a good MIC value since drugs could inhibit the bacterial growth by various pathways; then, the antibacterial activity would not be affected only by the inhibitory action on the FabH enzyme. To determine whether 2-pyrazoline derivatives inhibit the FabH enzyme and this inhibition results in its antibacterial activity, an experiment on the activity of FabH enzyme is also required. Finally, note that the virtual approach can not fully replace the conventional approach; therefore, a systematic structure exploration should be carried out to discover the highlight active compounds and to enriche the data of the structure-activity relationship of the 2-pyrazoline derivatives.
Conclusion
In short, our work successfully configures a docking model on FabH enzyme, which produces a statistically significant correlation between the docking score and the experimental MIC value. This molecular simulation also suggests the targeting of FabH enzyme as the mode of action of these 2-pyrazoline derivatives. This docking model is re-evaluated by both an external validation dataset and a series of newly synthesized 2-pyrazoline derivatives from this study. These four compounds (HP16-HP19) are first synthesized, and related data were not available in scifinder.cas.org (updated on May 30th, 2021). Taken together, our study provides a docking model which potentially predicts the activity against S. aureus of 2-pyrazoline derivatives. Nevertheless, limitation exists between the in silico simulation and the experimental approach. In fact, the docking model is restrictively built on FabH enzyme, whereas the biological activity is assessed on the whole bacteria. Consequently, it remains unclear whether the antibacterial activity is influenced by other biological processes. To confirm our hypothesis and consolidate the applicability of the docking model, FabH enzyme inhibition assays, however, need to be conducted in further research. Finally, the cytotoxic activity of selected 2-pyrazoline derivatives from our study positively expands the integrative database of this class of organic compounds.