
1. INTRODUCTION
Dengue virus (DENV) is a single-stranded flaviviridae RNA virus and responsible for causing the mosquito-borne infectious dengue disease mostly in tropical areas [1]. The disease manifestations range from mild dengue fever (DF) to the life-threatening dengue hemorrhagic fever (DHF) or dengue shock syndrome (DSS). Naturally, the virus is circulating in four major serotypes (DENV-1–4) and showing about 65–70 % sequence homology with each other [2]. These serotypes show the highest mutation rates among other flaviviruses [3, 4]. This has led to the formation of different lineages and genotypes within each serotype [5]. Accordingly, DENV-1 comprises of five genotypes (I–V); DENV-2 has six genotypes (Southeast Asian/American, Asian I, Asian II, Cosmopolitan, American, and Sylvatic); DENV-3 has four genotypes (I–IV); and DENV-4 also consists of four genotypes (I, II, III, Sylvatic) [6]. Genetic changes can predominantly affect the disease burden by changing genotypes and subsequent changes in disease virility. An example of that is the changes of DHF rates in Sri Lanka when the DENV-3 (genotype III) group A was replaced with group B viruses [7] . Another example is when the American DENV-2 was replaced with the more severe Southeast Asian DENV-2 and the associated changes following this event [8].
Vietnam is an endemic country of dengue; particularly Southern Vietnam being a hyper-endemic area with nearly ten-fold more dengue than Central Vietnam every year [9, 10]. According to the summary report of the Pasteur Institute in Ho Chi Minh City, in 2013-2015, the number of dengue cases had tended to increase from 33,626 cases in 2013 to 50,205 cases in 2015. DENV-4 has been found in each area of Vietnam (Southern, Northern, Central) with over 50 strains isolated and the data was available in GenBank [10] . In 2013, a large dengue outbreak occurred in central Vietnam resulting in 204,661 clinical cases, with DENV-4 being the most predominant serotype followed by DENV-1, DENV-3 and DENV-2, respectively [10]. Similarly, another outbreak was detected around the same period at Cat Ba Island with identified 192 cases [11]. The cases were attributed to DENV-3 serotype (genotype III), which presented a high homology with the DENV strains affecting the nearby city of Hanoi in the same year [11]. These finding suggested that the viruses were probably introduced to the islands from the mainland (likely Hanoi), hence, the prevention efforts should be focused mainly at the origin [11]. Additionally, DENV-3 (genotype III) was detected in cerebrospinal fluid of an accountant living in Hai Phong, which showed a close relationship with the aforementioned serotypes using phylogenetic analysis. Noteworthy to mention that DENV-3 (genotype III) was reported for the first time in Vietnam in 2013 while genotype II had been the one previously circulating in the country [12]. In 2011, DF epidemics were noted in Hanoi with 24 outbreak points hitting eight districts with serotypes DENV-1 and DENV-2 detected in human samples [13]. Back to 1998, a widespread epidemic occurred of DHF with the incidence rate of 438.98 cases per 100,000 population, representing a 152.4% increase compared to 1997 epidemic [9]. Although most prevalent serotype was DENV-3, other serotypes were noted as well but in lower percentages [9]. A recent study presented that DENV-1 strains in Northern Vietnam were most closely related to the dengue virus outbreak in Cambodia from 2006 to 2008 [14].
In this study, we aimed to identify a complete genome sequence analysis for all four types of DENV strains circulating in Southern Vietnam during 2014-2015. The identification of recent serotypes and genotypes of the strains will contribute in predicting the transmissibility and virulence of the virus responsible for epidemic.
2. MATERIALS AND METHOD
We employed Aedes albopictus (C6/36) cell line to grow viruses. C6/36 cells were cultured in 25cm2 flask (Nunc, Cat.No. 163371) with Dulbecco’s Modified Eagle Medium (Gibco, Cat.12100-061) supplemented with 10% Fetal Bovine Serum (FBS) (Gibco, Cat.10084-168) for growth at 28°C. Twenty-four strains of dengue viruses isolated in 2014-2015 and kept in −80°C storage were used to infect the cell lines. After the incubation period of 7 days at 28°C, infected cells were harvested and identified by direct fluorescent assay (DFA) as well as immunofluorescence assay (IFA) with the following monoclonal antibody: anti-Flavin conjugated fluorescein isothiocyanate (FITC), anti-dengue virus serotype 1-4 and anti-mouse IgG conjugated FITC (Sigma, F5262). The information about monoclonal antibody was provided by US CDC: DEN.1 Hawaii (D2-1F1-3); DEN.2 NGC (3H5-1-21); DEN.3 H87 (5D4-11-24); and DEN.4 H241 (1H10-6-7).
Study samples were collected by Centers for Disease Control (CDC) in 20 provinces in the Southern Region. The agency responsible for isolation and sequencing is the Pasteur Institute in Ho Chi Minh City, Vietnam.
RNA from dengue viruses of which serotypes had been identified (1 to 4) was extracted using the QIAampR Viral RNA Mini Kit (Qiagen 52906). Full genome of the virus strains was identified using the Superscript III Reverse Transcriptase kit (Invitrogen, Cat. 18080-051) and the Platinum Taq DNA polymerase (Invitrogen, Cat. 10966-018). Brieflt, we used Superscript III RT and random primer to synthesize cDNA. Then, we performed PCR to collect full genome by Plantinium Taq DNA polymerase (Invitrogen, Cat. 10966-018) with primer in Supplementary Table S1. Specifically, for the Superscript III Reverse Transcriptase kit, the reaction mixture recipe was 1 μL random primer, 1 μL dNTP, 3 μL diethyl pyrocarbonate (DEPC)-treated water, 2 μL buffer 10X, 4 μL Mg2+, 2 μL DDT, 1 μL RI, 1 μL Enzyme Reverse Transcriptase and 5 μL RNA target with the following thermal cycle: 25°C for 10 minutes, followed by 50°C for 50 minutes, then 85°C for 5 minutes, and finally 4°C for 60 minutes. For Plantinium Taq DNA polymerase (Invitrogen, Cat. 10966-018), the reaction mixture recipe was 2.5 μL Buffer 10X, 1 μL Mg2+, 0.5 μL dNTP, 0.5 μL for each forward primer and reverse primer, 0.1 μL enzyme, 17.4 μL DEPC and 2.5 μL cDNA. DNA polymerase activation was followed by 35 amplification cycles of 30 seconds at 94°C, then 30 seconds at 55°C and 3 minutes at 72°C.
The genome virus was purified using the QIAquick ® PCR Purification (Qiagen 21480) and QIAquick ® Gel Extraction kit (Qiagen 28704). DNA fragments from 100bp to 10kb were purified from primers, nucleotides, polymerases, and salts by the QIAquick spin column under the effect of centrifugation.
DNA concentration was quantified by Qubit fluorometer. The fluorescent dyes were associated with specific target molecules. Calibration curve was used to generate the quantitation results. Later, the DNA concentration was diluted to 0.2 ng/μL, then the DNA was ligated and indexed by PCR reaction with the following reaction mixture: 25 μL DNA, 15 μLNPM, 5 μL Index 1 and 5 μL Index 2. The thermal cycle was 72°C for 3 minutes, followed by 95°C for 30 seconds, then 12 amplification cycles of 10 seconds at 95°C, 30 seconds at 55°C and 30 seconds at 72°C, then 72°C for 5 minutes and finally 4°C for 60 minutes.
After being attached with the index, the products were purified by AMPure XP beads kit (Beckman Coulter A63880). The genome of dengue viruses was sequenced using the Next-Generation Sequencing (NGS)-Illumina method in MiSeq system. Briefly, the DNA adhered to the adapter would be attached to the flowcell. Each small segment on the flowcell was synthesized to form a cluster to amplify the read signal. Each run provided a line of A, T, G, or C, which would be attached to the blocker. The signal from the attached unit would be received, analyzed and processed [15]. Finally, the sequencing results were analyzed by BLAST program [16]. The phylogenetic analyses and estimates of transition capacity between DENV nucleotides were conducted using MEGA7 version 7 (http://www.megasoftware.net/). The sequences were purified by CLC Genomics Workbench 10.1.1 software and aligned by the maximum likelihood estimation method with bootstrap analysis of 100 repetitions and reliability value greater than 70%. The analysis begins with a specialized tree created from the input data. The original tree branches were swapped until the tree with the highest likelihood score was obtained [17]. All reference sequences of different geographical regions were retrieved from GeneBank and used for genome analysis to create phylogenetic trees (Supplementary Table S2).
3. RESULTS
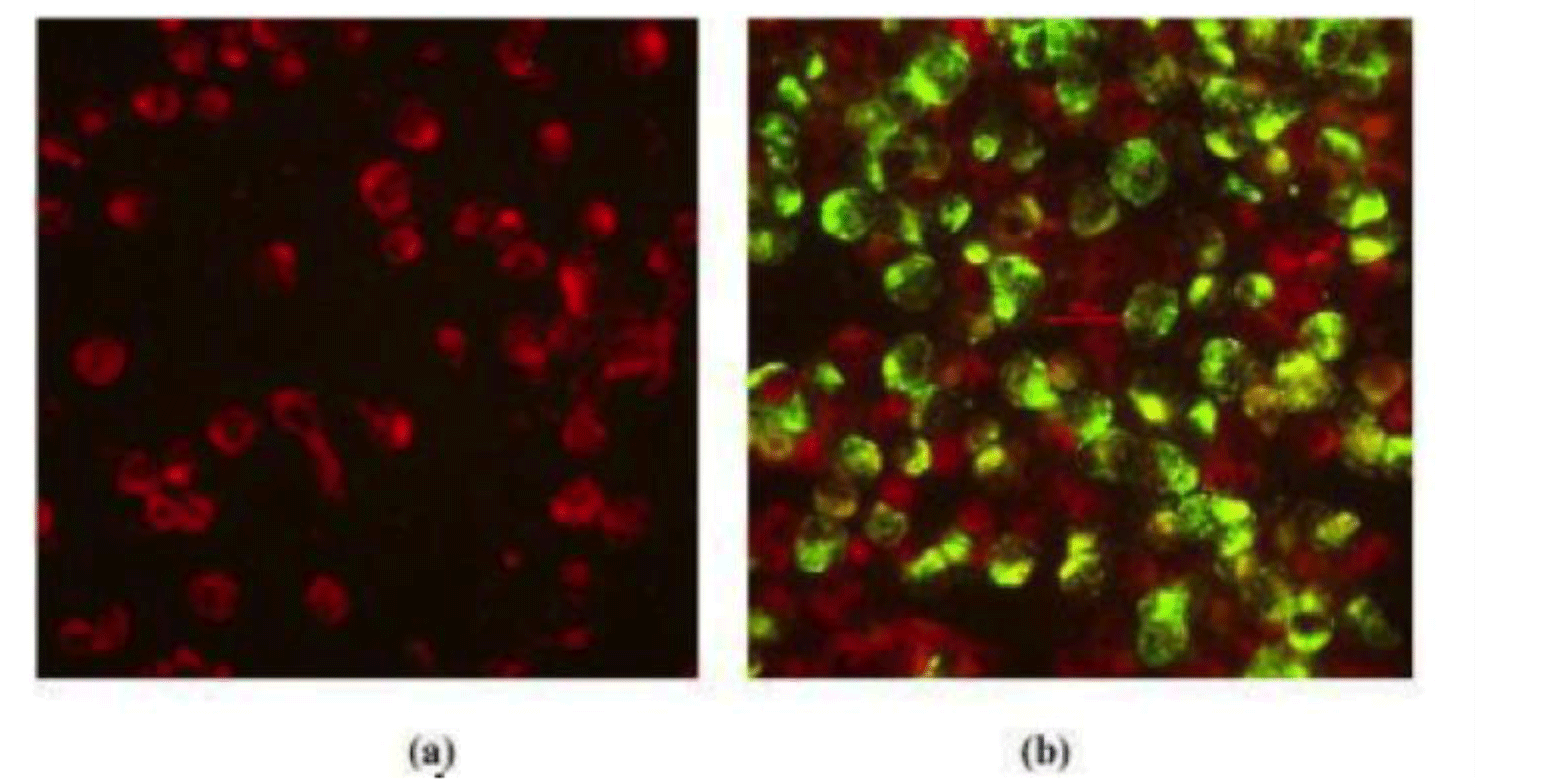
The results of virus isolation were presented in Table 1. Of 24 strains, we identified 20 positive strains, including 4 strains of DENV-1, 6 strains of DENV-2, 5 strains of DENV-3 and 5 strains of DENV-4. The results of IFA under fluorescence microscope were presented in Fig. 1. The positive samples after virus isolation were sequenced. The DENV genome was about 11kb in length. From 20 sequenced samples, after purification by CLC Genomics Workbench, 17 strains had appropriate quality for sequencing and further genotype analysis (Supplementary Table S2). Of these 17 DENV isolates, 4 (23.53%) were DENV-1, 3 (17.65%) were DENV-2, 5 (29.41%) were DENV-3, and 5 (29.41%) DENV-4 was identified in our study. Most of them distributed across the Southern Vietnam regions including Dong Thap, Ba Ria-Vung Tau, Binh Duong, Soc Trang, Ben Tre, Tay Ninh, Ho Chi Minh City, Dong Nai (Table 1).
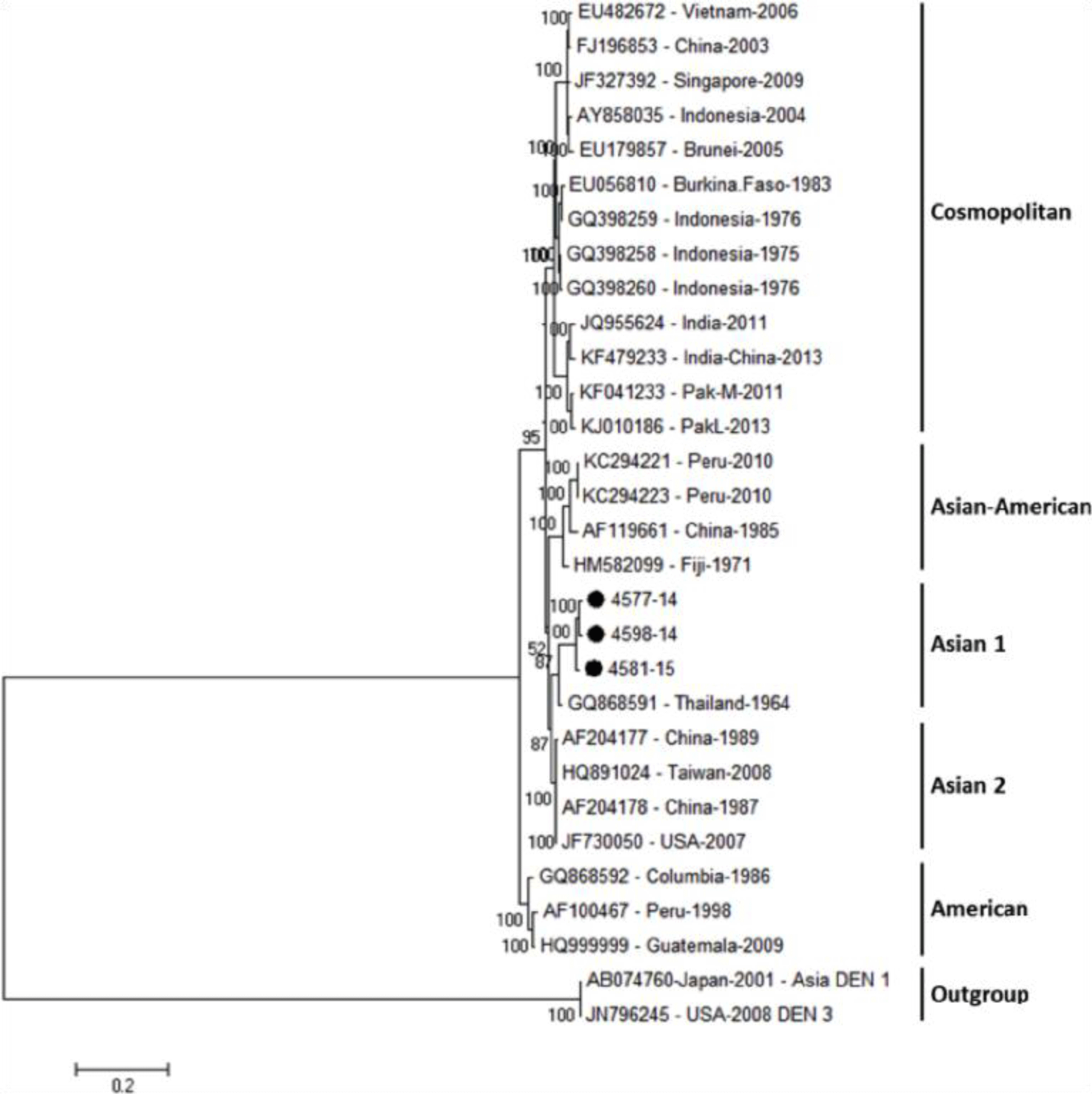
Phylogenetic tree revealed that DENV-1 strains were divided into three main genotypes according to the geographic area, including Asia, South Pacific and America/Africa, in which four genome sequences of DENV-1 isolates belong to small subgroup of Asia genotype and genetically close to previous strains from Vietnam, Thailand, China and Sri Lanka (Fig. 2). For DENV-2, the phylogenetic tree showed three DENV-2 isolates were belong to Asian 1 group and genetically closest to the Thailand strain (Fig. 3).

Regarding DENV-3, the phylogenetic analysis reported that five strains isolated from our study were identified to group of Asian 2 genotype and genetically close to previous strains from Vietnam, Cambodia, Thailand and Singapore (Fig. 4). For DENV-4, the phylogenetic analysis demonstrated that five DENV-4 strains isolated from our study were identified as Asia 1 genotype. DENV-4 isolates were genetically close to other previous isolates from Thailand and Brazil (Fig. 5).


The transition capacity among nucleotides of four DENV was presented in Table 2. The result of analysis of DENV-1 showed that the highest ratio mutation was from C to T at 31.99% and the lowest ratio mutation was from G to C at 0.67%. Similarly, in DENV-2, the ratio mutation C to T was highest at 27.54% meanwhile from A to C was lowest at 1.61%. In DENV-3, the highest ratio mutation C to T was 33.87% whereas from G to C was 0.92 % and in DENV-4, the ratio mutation C to T was highest at 29.87%, while G to T was 0.5 %, respectively.
Overall, the result of analysis revealed that the probability of conversion from C to T was the highest conversion rate among all four types of DENV-1, -2, -3, -4 serotypes. For comparison at the level of amino acids, the percentage difference in each pair of amino acid sequences between the four strains ranged from 0 to 1.03% (Supplementary Table S3).
4. DISCUSSION
In this study, we identified 17 complete DENV genome sequences, including 4 strains of DENV-1, 3 strains of DENV-2, 5 strains of DENV-3 and 5 strains of DENV-4 from patients with dengue during 2014-2015 in Southern Vietnam. Based on maximum likelihood method, the phylogenetic trees were constructed to detect any changes in the circulating strains of dengue in Southern Vietnam. Maximum likelihood method was considered as the most suitable fit for phylogenetic trees with large number of taxa and long sequence strings for viral phylogeny with its added bootstrap technique and high accuracy that we used to determine the branching patterns [18]. This information will help health authorities identify and take the necessary measure to prevent another epidemic.
DENV-1 was divided into the following genotypes: sylvatic/Malaysia, America/Africa, South Pacific, Asia, and Thailand [18]. Our phylogenetic analysis of four strains of DENV-1 revealed that the circulating strains belong mainly to Asian lineage and Thailand group. It was also related to both South Pacific and America/Africa. This demonstrates the diversity of the strains isolated from our cases. This was consisted with a study performed in Taiwan where they isolated DENV-1 from travelers returning from Vietnam. They observed this diversity in the strains of circulating genotypes in Vietnam [19]. Another study isolated dengue virus from 81 Vietnamese patients, in which, Rabaa et al. found that the circulating DENV-1 lineage in Southern Vietnam had come from Cambodia which contracted this lineage from Thailand [20]. This explained our tree results, which has shown genetic proximity to Thailand strains. Another study supported our finding that the DENV-1 strains were mainly distributed from Thailand and Indonesia [21]. Moreover, Mizuno et al. also proved that Vietnamese strain was the origin for strains circulating in China, Cambodia and Malaysia. Strains circulating in Japan and Korea were also found to be from South East Asia [22]. For its relation to Americas/African strain, a phylogenetic analysis revealed that the circulating strains in Caribbean and Americas was introduced from South Asia, mainly Thailand, which may explain the phylogenetic relationship between the strains [21].
Identification of the phylogenetic relations to other genotypes of DENV-1 will predict the incidence and severity of DENV-1 epidemic in Southern Vietnam [23]. It is well-known that American/African genotype has higher likelihood to cause DHF and more severe form of disease [18]. These strains also had a high potential to cause an epidemic [18, 23]. Similarly, DENV-1 also reached the dominant serotype in the outbreak of Myanmar in 2013, in which, the primary DENV infection remained at high level among the severe dengue cases [24].
In addition, the analysis revealed that three isolates of DENV-2 sequences in Vietnam were concentrated in a subgroup of the Asian genotype which along with the 1964 Thailand strain, are called Asian 1. A phylogenetic analysis of 273 strains of Asian lineage was divided into two groups; one comprised all strains from Thailand and China strains isolated during 1985–2001 and the other included Vietnam, Thailand and Cambodia from 2001 to 2008 [25]. In our analysis, our strains were related to older strains of the 1964 Thailand strain. It was demonstrated that the parent strain of modern strains in Vietnam is from Thailand [25]. It was also found that the strains in nearby regions are from Thailand. These findings were also supported by findings from a study in Central Vietnam during the outbreak of 2010-2012 [26]. Moreover, our results were consistent with another study that studied the evolution of DENV-2 in Vietnam [27]. They found that Asian I lineage had totally replaced the predominant Asian/American lineage since 2003 [27]. This was highly important since it was found that Asian strains were more related to development of sever forms of disease unlike other strains [23, 28]. It was able to have persistent high blood viremia in dengue patients. It was also found that genotypes of DENV-2 had high transmissibility, signifying its epidemics [18, 23]. A most recent study reported that DENV-2 isolated from the 2017 dengue outbreak in Northern Vietnam originated from India in 2006 [29].
Other two serotypes extracted from our patients were DENV-3 and DENV-4. DENV-3 isolates from our study were found as genetically close to previous strains from Vietnam, Cambodia, Thailand and Singapore. This finding was in agreement with results from Podder et al. [30] and Shu et al. [31] on the E gene region. Phylogenetic analysis of DENV-3 using E gene revealed four subtypes based on geographical distribution including Americas, Indian subcontinent, Thailand and Southeast Asia/South Pacific [18]. This classification was useful to identify the virulence and epidemic potential for the circulating serotype across the years in each country. The year of 1990 was considered as the cut-off year for the circulating DENV-3 in America, before 1990, only American genotype was the one circulating with low incidence of new cases and only DF was diagnosed [32, 33]. Unlike after 1990, Southeast Asian serotypes were associated with high epidemic and more diagnosis of DHF; Indian type was also found after 1990 in Americas and Asia and was correlated to severity of the disease [34, 35].
In case of DENV-4, the rate of incidence of epidemic from this serotype was less predominant than other serotypes imposing an obstacle for its classification into well-established genotypes [18]. Besides, our DENV-4 isolates were proved to genetically close to other previous isolates from Thailand and Brazil. This is consistent with the hypothesis that the presence of the Americas is due to the spread of these strains from Asia to America [36]. After the discovery of full sequence of E gene, only three genotypes were reported with only one type found to be circulating along many continents. Still, more efforts are needed to classify this serotype [36, 37].
The result of analysis revealed that the probability of conversion from C to T was the highest conversion rate among all four types of DENV-1, -2, -3, -4 serotypes. Based on the wobble theory [38], the transition from C to T was more likely to cause significant mutations on the protein surface such as Ser transfer to Phe and Leu, or Leu to Pro. These findings could play a key role in the genetic evolution. In addition, the percentage difference in each pair of amino acid sequences between the four strains ranged from 0 to 1.03% (Supplementary Table S3). However, the degree of amino acid sequence difference is always lower than that at the nucleotide level, indicating that most mutations occur in the third-base of the coding codon. There is almost no change at the amino acid level, so this result shows that most of these isolated viruses are in the stage of accumulation of mutations.
Conclusion
In conclusion, our study describes, for the first time, the genetic diversity of all four serotypes of DENV in Southern Vietnam during 2014-2015. We demonstrated that DENV is hyperendemic in Southern Vietnam, rendering co-infection between serotypes and genotypes possible. We observed the co-circulation of multiple genotypes within DENV-1 to DENV-4; four DENV-1 isolates belong to Asia genotype, three DENV-2 strains were concentrated in a subgroup of Asian 1 genotype, five DENV-3 isolates were identified as belonged to Asian 2 genotype and five DENV-4 isolates were found as belong to Asia 1 genotype. These findings highlighted the geographic distribution and dynamic transmission of DENV strains occurred in Southern Vietnam. The transition capacity between the nucleotide among all four types of DENV-1, 2, 3, 4 serotypes also suggested that the probability of conversion from C to T was the highest conversion rate, which implied genetic evolution of DENV strains.
