
1. INTRODUCTION
Patients with systemic lupus erythematosus (SLE) are affected by various antibodies leading to a range of complications, which include a distinctive, widespread blister eruption characterized by a dermatitis herpetiformis-like histology, linear and/or granular basement membrane zone (BMZ) antibody deposition typically reacting with type VII collagen, and a striking response to dapsone. This condition constitutes bullous systemic lupus erythematosus (BSLE), which occurs in less than 5% of patients with SLE [1] and accounts for 2-3% of autoimmune subepidermal blistering diseases [2-4]. B SLE tends to affect young women more often than men [1,5] with the estimated incidence does not exceed 0.2 cases per million per year [6], usually in their second and third decades of life, and is associated with lupus nephritis in 50% of cases [7]. The diagnosis of this disease primarily requires the diagnosis of SLE according to the American College of Rheumatology (ACR) criteria in the combination of clinical features, histological evidence, characteristic direct immunofluorescence and/or indirect immunofluorescence microscopy findings as well as other immunologic tests to determine the presence of specific autoantibodies [1,7-9]. Patients usually present with an acute, widespread, tense vesiculobullous eruption that mostly affects sun-exposed areas [1,7,10]. A dramatic clinical response to dapsone is characteristic and supports its use as the initial treatment of choice for B SLE [1,11-14]. Herein, we report a case of BSLE in a 16-year-old Vietnamese girl, who demonstrated an acquired vesiculobullous eruption associated with nephrotic syndrome and fulfilled the European League Against Rheumatism (EULAR)/ACR 2019 classification criteria of SLE [15]. We also briefly review clinical, histopathological, and immunological characteristics of BSLE for an overall picture of diagnosis.
2. CASE REPORT
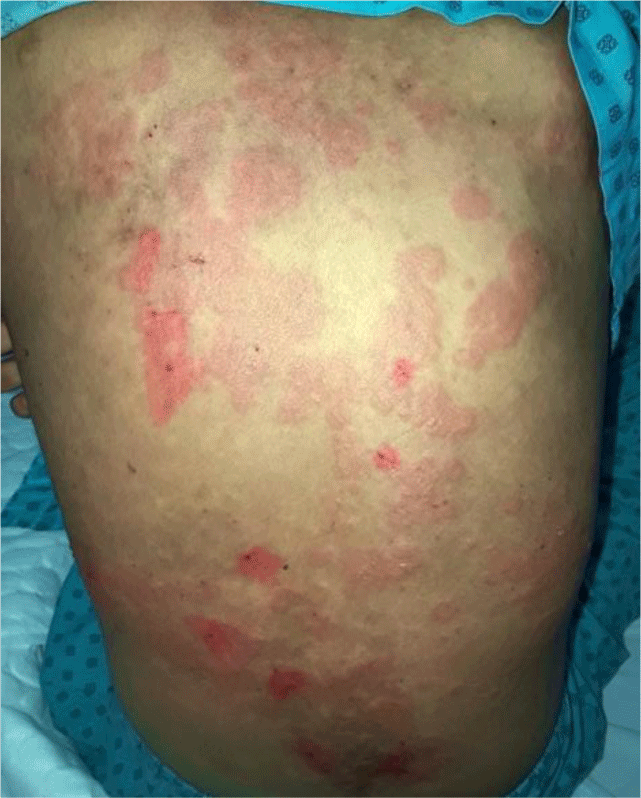
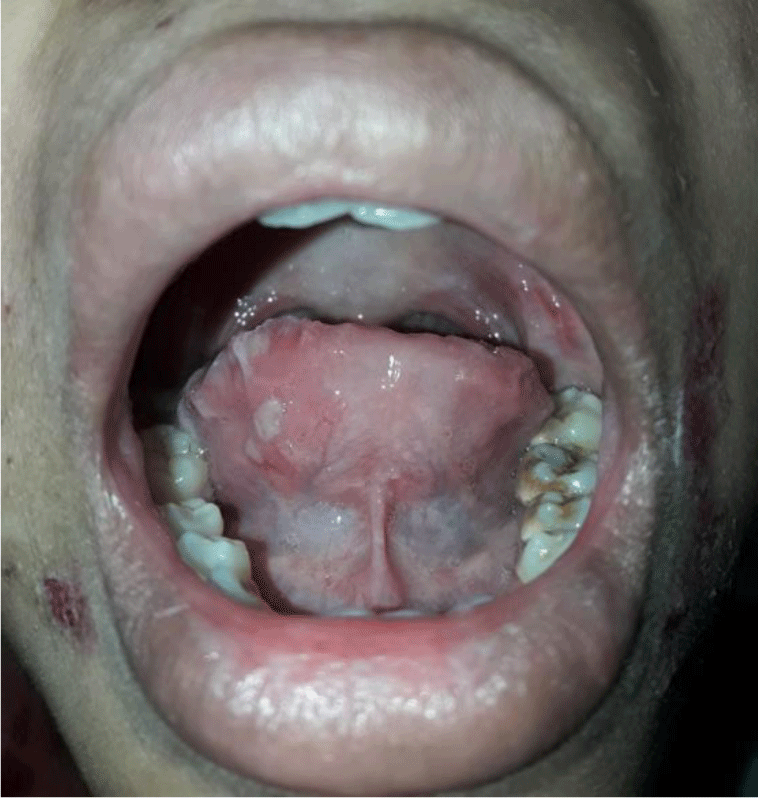
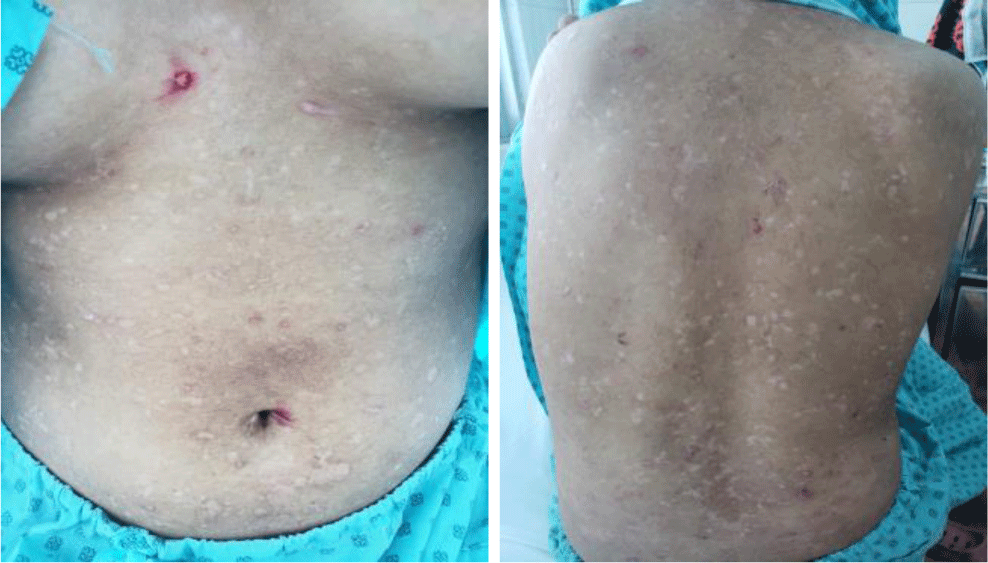
A 16-year-old girl presented with a one-month history of a generalized skin vesiculobullous eruption preceded by two weeks of painful oral mucosal lesions. The patient had a history of photosensitivity, but she had not experienced fever, arthralgias and myalgias. Physical examination at admission showed blood pressure of 100/60 mmHg, respiratory rate of 17/min, and heart rate of 76/min. The patient’s bodyweight and height were 44 kg and 151 cm, respectively. The body mass index value (19.30 kg/m2) was within the normal range for Asian populations. Body temperature was 36.7°C at presentation and fluctuated from 36.3°C to 37.1°C during the patient’s hospital stay. No abnormalities in the heart and lungs were noted. Dermatological examination showed numerous tense, clear fluid-filled vesicles and bullae overlying erythematous plaques (Figure 1A) that interpersed with several rounded, sharp-edged erosions (Figure 1B). The blistering displayed the predilection for the face, neck, chest, abdomen and bilateral inner thigh areas and spared palms and soles (Figure 2). Vesicles and bullae were dome-shaped and measured from 1 milimeter up to 2 centimeters in diameter (Figure 1A) that healed with post - inflammatory hypopigmentation without scarring. In addition to blisters, there were erythematous plaques sized 1 to 4 centimeters in diameter with annular configuration arising on the back skin (Figure 3). Well-circumscribed, rounded erosions were observed on the tongue and the buccal mucosa (Figure 4).
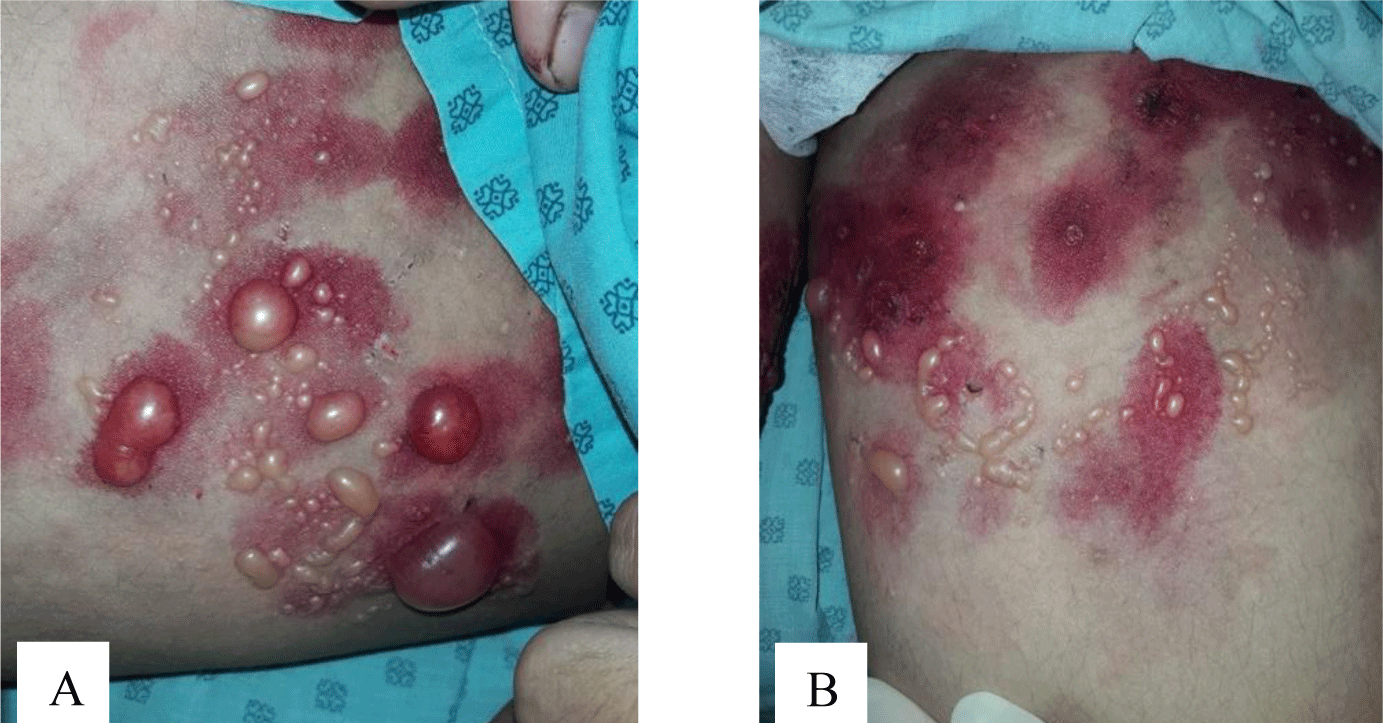
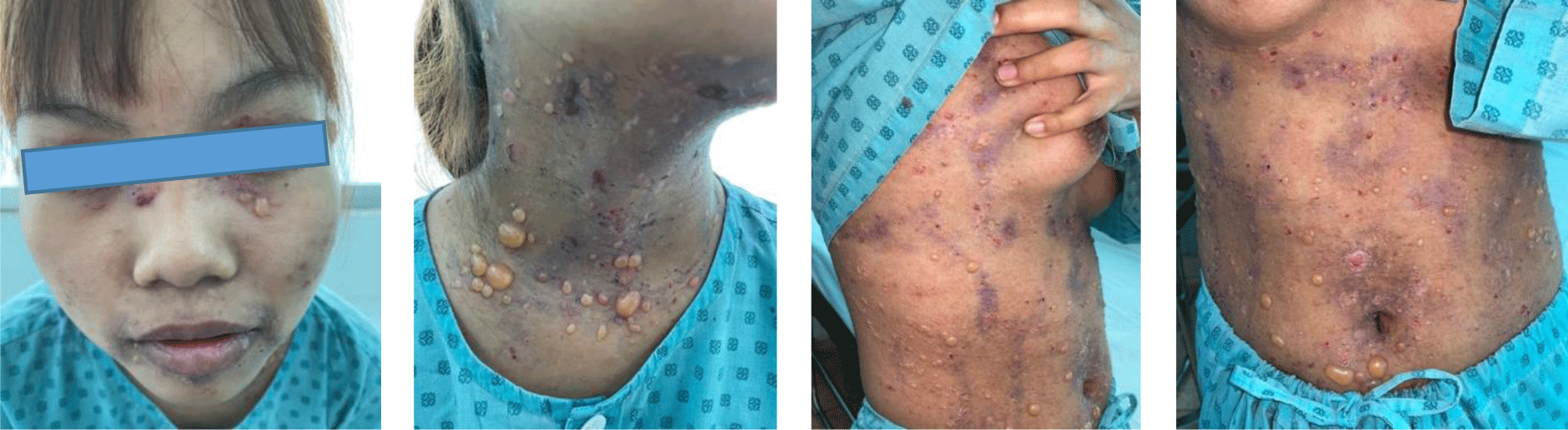
Annular distribution of vesicles sparsely presented (Figure 1B) but rossette-like pattern and “string of pearls” sign of vesicles were not found. Mechanobullous lesions were not seen. Nikolsky’s sign was negative. Malar rash and discoid rash were absent. Pitting edema over the legs developed after presenting to the hospital.
The patient’s complete blood count revealed a microcytic hypochromic anemia, an increased white blood cell count and a normal platelet count. The initial dipstick analysis displayed significant proteinuria without hematuria. The urinary total protein level rose to 3.47 g/24h. Serologic tests showed positive antinuclear antibody (ANA) but negative antidouble-stranded DNA (anti-dsDNA). Complement component assays revealed low levels of C3 and C4.
A saucerization biopsy specimen was obtained from the edge of an intact blister on the patient’s abdominal skin that contained both portions of the bullae and nonbullous skin. This specimen was fixed in 10% neutral buffered formalin, then processed, and embedded in paraffin wax according to standard histological procedures. Four-micron sections were cut for hematoxylin and eosin (H&E) staining.
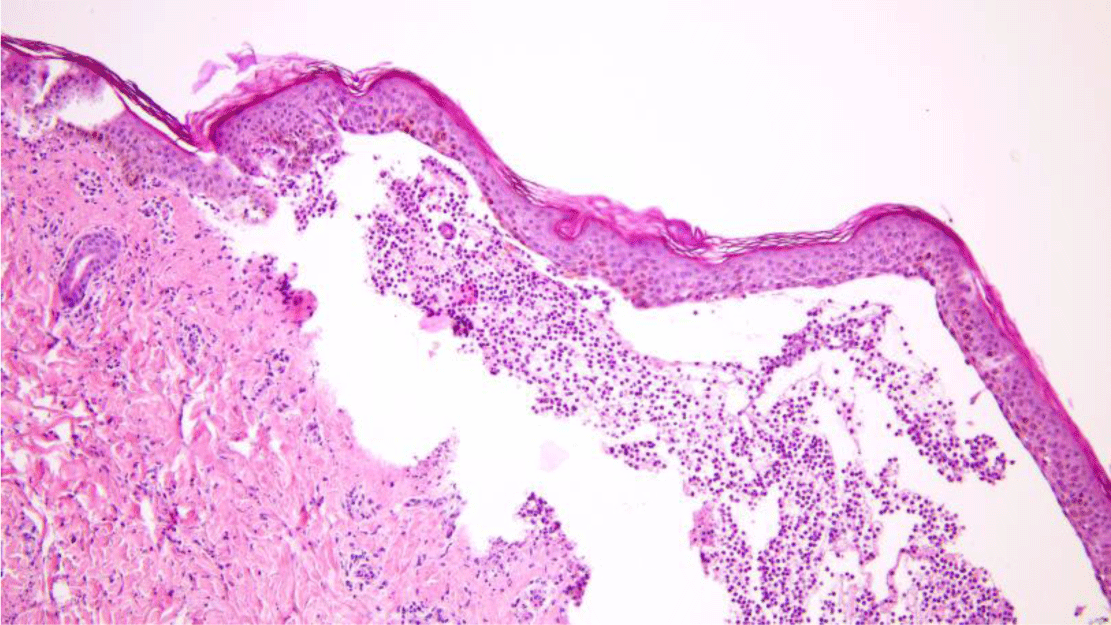
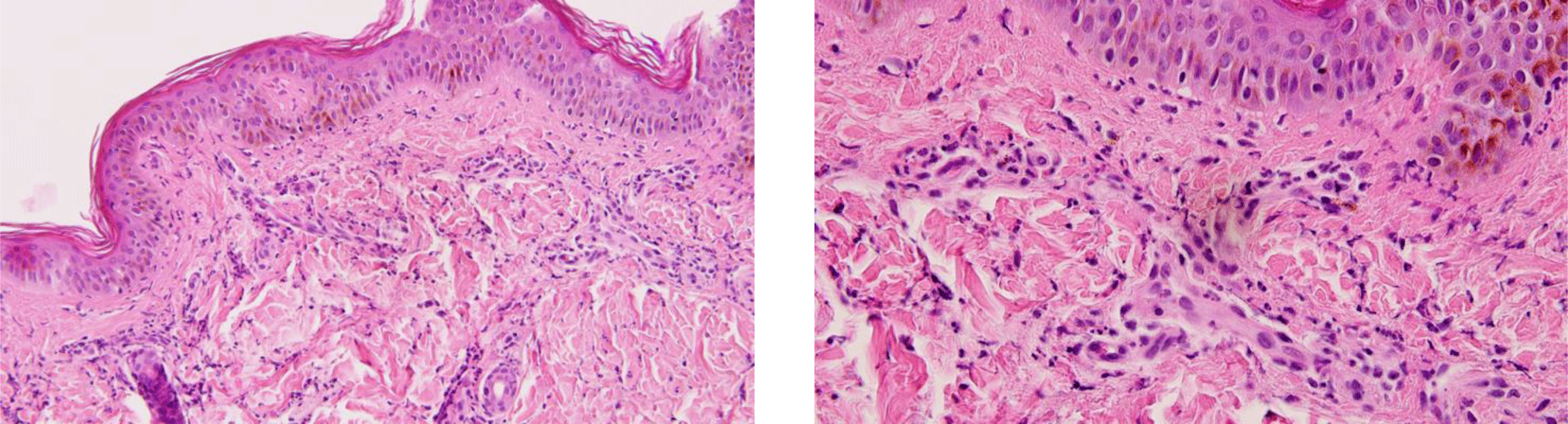
Another saucerization biopsy specimen for DIF was taken from nonbullous erythematous skin within 1 cm of a bulla on the abdomen, which was placed on normal saline-soaked gauze and transported to the laboratory within 24 hours. This specimen was embedded in optimal cutting temperature compound, frozen in a cryostat at -20°C. Six 4-μm sections of the specimen were taken onto charged slides and air dried for 15 minutes. All six slides were then placed in acetone at -20°C to fix for 10 minutes, let airdry for 5 minutes, and permeabilized with 0.3% triton x-100 in Phosphate Buffered Saline (PBS) for 10 minutes before being rinsed in PBS. These slides with the exception of the negative control one were incubated with fluorescence isothiocyanate labeled primary antibody (polyclonal rabbit anti-human fibrinogen/FITC, polyclonal rabbit anti-human IgG/FITC, polyclonal rabbit anti-human IgA/FITC, polyclonal rabbit antihuman IgM/FITC and polyclonal rabbit anti-human C3c complement/FITC were applied for separated slides except the negative control slide) at 37°C for 1 hour. All slides, which included the negative control one, were then rinsed 3 times in PBS for 5 minutes each to wash off unreacted antibodies before being mounted with a drop of buffered glycerin. Histopathologic examination of the lesional skin biopsy showed subepidermal vesicle containing fibrin and abundant neutrophils along with scattered eosinophils, lymphocytes and histiocytes (Figure 5). The upper dermis contained a perivascular mixed inflammatory cell infiltration which predominantly consisted of neutrophils, occasional eosinophils and lymphocytes (Figure 6). Dermal papillary microabscesses and the features of a leukocytoclastic vasculitis were absent.
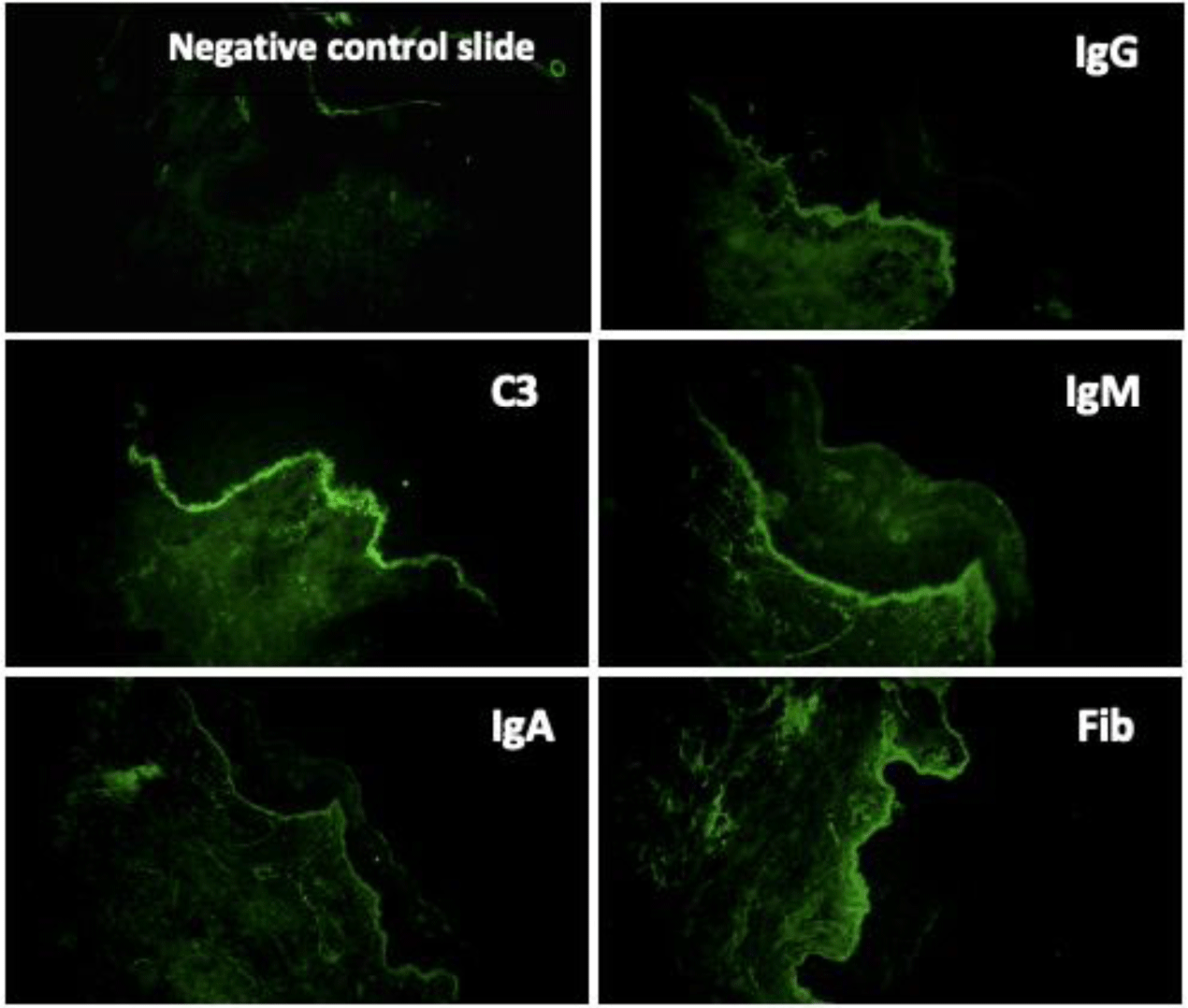
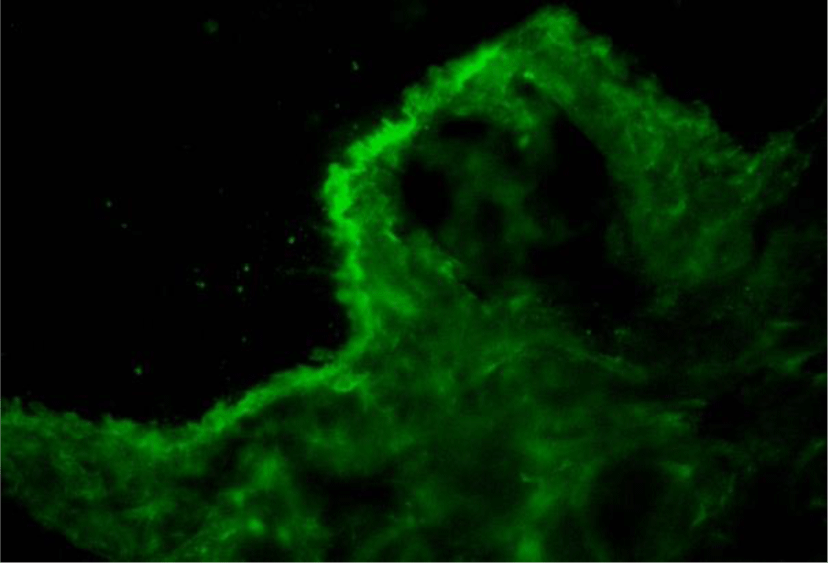
Direct immunofluorescence of perilesional skin biopsy demonstrated the linear deposition of IgG, IgA, IgM, C3 and fibrinogen at BMZ (Figure 7). The immunoglobulin deposition pattern displayed u-serrated type (Figure 8) with the predominant staining intensity of IgG in comparison with C3 and IgA. The final diagnosis of BSLE was made.
The patient was treated with a high-dose oral glucocorticoid scheme. Treatment initiated with methylprednisolone 1 mg/kg/day for two weeks, but skin lesions showed negligible improvement. Then higher dose of methylprednisolone at 1.5 mg/kg daily was administered for the following 12 days and resulted in significant clinical recovery with cessation of new blister formation and healing of existing lesions (Figure 9). However, with persistent significant proteinuria (without hematuria) from urine dipstick analysis, she was transferred to another general hospital for continuing treatment of renal complications.
3. DISCUSSION
BSLE occurs in less than 5% of patients with SLE [1] and accounts for 2-3% of cases of autoimmune subepidermal blistering disease [2-4] with the estimated incidence does not exceed 0.2 cases per million per year [6]. BSLE presents more frequently in women than men [1,5], especially of African descent, usually in the second to fourth decade [1,5,14]. Our case occured in an aldolescent girl first diagnosed with BSLE at the age of 16.
At the time of presentation, as this case manifested mostly non-scarring, tense, dome-shaped, clear fluid -filled vesicles and bullae measuring up to centimeters in diameter and distributing all over the body, the initial diagnosis was bullous pemphigoid (BP), which is clinically indistinguishable from pemphigoid-like epidermolysis bullosa acquisita (EBA). Besides, because the annular configuration of vesicles was sparsely found, linear IgA dermatosis (LAD) was considered as another differential diagnosis. In addition, pitting edema of legs and the history of photosensitivity suggest the diagnosis of BSLE. In regard to skin manifestation, BSLE is characterized by widespread, tense, occasionally pruritic vesiculobullous eruption that is not limited to sun-exposed areas [1,8,10]. Vesicles and bullae typically show a predilection for the face, supraclavicular regions, upper trunk, upper extremities, vermillion border and oral mucosa [1,7,10,14]. The vesiculobullous eruption may arise on an erythematous background or normal skin or, less commonly, urticaria [1,10]. The blister cavity usually contains clear fluid, or sometimes, hemorrhagic fluid [1,14]. Postinflammatory hypopigmentation is common but scarring formation is typically absent. Skin lesions of our patient had quite typical features of BSLE and distributed on both sun-exposed and non-sun exposed areas with the predilection for the face, neck, chest, abdomen and bilateral inner thigh areas. Unlike blisters of classic EBA, vesicles and bullae of this patient spared palms, soles as well as other trauma-susceptible sites and healed with hypopigmentation without scarring and millia. However, the clinical presentation of pemphigoid-like EBA is closely resemble BSLE [1]. Being different from LAD, in this case, annular distribution, rossette-like pattern and “string of pearls” sign of vesicles did not present. As patients with B SLE do not usually develop characteristic cutaneous manifestations of SLE [1,7,10,14], it is reasonable that malar rash and discoid rash were absent in our patient.
Lupus renal involvement is one of the most common and serious complication of SLE, which affects 60% of SLE patients at 5 years post-diagnosis [16]. Comparably, a multicenter retrospective study on BSLE by de Risi-Pugliese et al. reported that lupus nephritis occurred in 50% of the BSLE cases, mostly class III or IV, according to the International Society of Nephrology/Renal Pathology Society (ISN/RPS) classification [7]. The manifestation of renal disease in SLE is variable, ranging from no symptoms, trace proteinuria to nephrotic syndrome and acute nephritic syndrome [16]. The presentation of lupus renal disease in this case was nephrotic-ranged proteinuria without hematuria, which is a clinical evidence of lupus nephritis. As the patient’s renal biopsy was not obtained, the diagnosis of lupus nephritis could not be confirmed and further classified. The nephrotic syndrome in this patient seems to arise after blistering eruption as pitting edema of bilateral legs had developed after presenting to the hospital. In a study of 10 patients with BSLE, Chanprapaph et al. reported four cases had bullous lesions as the initial manifestation of SLE and three of them had subsequent proteinuria [5]. Fujimoto et al. also reported a woman with BSLE presented with vesiculobullous eruption, who had been diagnosed with BP before the onset of marked proteinuria and rapidly progressive glomerulonephritis [17]. Therefore, vesiculobullous eruptions can arise as the onset of SLE, so it should be evaluated with cautiousness, and a carefully systemic examination is required to detect and timely prevent further complications that may occur.
Currently, the diagnosis criteria of BSLE include five elements [12]: (1) a diagnosis of SLE by American College of Rheumatology (ACR) criteria; (2) an acquired vesiculobullous eruption arising upon but not confined to sun- exposed skin; (3) histological evidence of a subepidermal blister with predominantly neutrophilic infiltration; (4) DIF microscopy demonstrating IgG with and without IgA and IgM deposits at the BMZ and (5) evidence of antibodies to type VII collagen via DIF or indirect immunofluorescence (IIF) on salt-split skin, immunoblotting, immunoprecipitation, enzyme-linked immunosorbent assay (ELISA), or immunoelectron microscopy. According to these , BSLE can be of three following types: type I satisfies all five criteria, whereas fulfilling only the first 4 criteria allows us to diagnose type 2 (undetermined location of antigen or dermal antigen other than type VII collagen) and type 3 (epidermal antigen) [12] . Serologic tests of our case showed positive ANA, which met the entry criterion for SLE. Additionally, for additive criteria, this patient presented painful oral ulcers and nephrotic range proteinuria as clinical criteria and the reduction of both C3 and C4 as an immunology criterion. On the basis of these, she was diagnosed with SLE according to 2019 EULA/ACR criteria [15].
The classical histopathology of BSLE demonstrates a subepidermal vesicle with papillary-tip neutrophilic microabscesses closely similar to histopathology of dermatitis herpetiformis (DH) [1,10,14,18]. However, the 128-patient retrospective study of de Rissi et al. had reported that dermal papillary microabscesses were seen in just 27% of BSLE cases. In fact, there were two forms of histopathology of BSLE were reported in the literature, one was dermatitis herpetiformis-like supepidermal blistering with neutrophilic microabscesses at tips of dermal papillae [1,10,14,18,19] and the other was subepidermal blistering with neutrophils evenly lining along the dermal-epidermal junction in a band-like pattern [5,7,9,14,20]. In our case, the lesional biopsy showed subepidermal vesicle containing a lot of neutrophils. Microabscesses at tips of dermal papillae consistent with DH were not present in this patient’ s lesion biopsy. Nonetheless, the alignment of neutrophils with interspersed leukocytoclasis along junctional zone was observed. Additionally, though the features of leukocytoclastic vasculitis were absent, a perivascular mixed inflammatory cell infiltration which predominantly consisted of neutrophils was prominent in this case. De Rissi et al. reported 9% of cases with BSLE displayed histological signs of vasculitis in lesional biopsies [7]. Vasculitis changes in BSLE can be perivascular inflammatory infiltration in the upper and middle dermis or prominent leukocytoclastic vasculitis [1,13,14,21].
With regard to immunopathologic features of BSLE, perilesional and clinically uninvolved skin typically demonstrate deposits of all major immunoglobulins (IgG, IgA, IgM) and complement components along the BMZ under DIF tests. The study of 128 cases with BSLE by de Rissi et al. reported that DIF was positive at the BMZ in 98% of BSLE cases, deposits of IgG was present in 91% of cases, IgA in 72% of cases, IgM in 68% of cases and C3 in 67% of cases. An other review of 30 BSLE cases by Flemming et al. [11] reported IgG was present in 93% of cases, C3 in 77% of cases and IgM and IgA in about 70% of cases [11]. Complement components are more commonly detected in lesional skin and much more rarely observed in clinically unaffected skin [1,13,14]. In BSLE, the two most common patterns of immunoreactant deposition at BMZ are granular and linear. According to Camisa and Sharma [8], if there is a linear pattern of immunoglobulin deposition, to distinguish from BP, immunoelectron microscopy should be done to identify the immunoreactants on or below the lamina densa. In our case, DIF of perilesional skin biopsy demonstrated the deposition of IgG, IgA, IgM, C3 and fibrinogen at BMZ. The IgG deposition showed a linear type. However, this linear deposition displayed a u-serrated pattern, which indicated that immune reactants located below the lamina densa, codistributed with type VII collagen. Type VII collagen is the major component of anchoring fibrils located beneath the lamina densa and known as the “sub-lamina densa fibrillar zone” [22,23], which attaches the epidermis to the dermis [22,23]. Autoantibodies to type VII collagen are also encountered in EBA. Current immunologic criteria for the diagnosis of BSLE are not different from those of EBA [1]. Moreover, the mucosa and skin manifestation of BSLE is also confused with EBA in many cases, especially with pemphigoid-like EBA. However, in most cases, the diagnosis of BSLE can be correctly established with the combination of clinical picture, histologic and immunologic features along with the fulfillment of ACR criteria for SLE. In addition, a remarkable feature to distinguish BSLE from EBA is the response to dapsone. Dapsone is effective in about 90% of BSLE patients and result in clinical improvement within days to a few weeks [1,5,7,14], while most EBA required systemic corticosteroids or immunosuppressors to control the clinical progression [1,24]. Being afraid of hematologic side effects of dapsone as our patient presented a moderately microcytic hypochromic anemia, she was treated with high-dose prednisolone alone and show gradually clinical improvement within 20 days. De Rissi reported corticosteroids as monotherapy were efficacious in 34% of cases. High-dose corticosteroids often used for the treatment of systemic symptoms of SLE are relatively ineffective in treating the cutaneous manifestation [14]. However, in BSLE occurring in the setting of SLE flares with renal involvement as our case, treatment with high-dose corticosteroids is a suitable choice [13].
Conclusion
It is difficult to make the correct diagnosis of BSLE due to clinical confusions with BP, LAD and EAB, histologic similarities to DH and EBA and a closely immunologic resemblance to EBA. The diagnosis of BSLE is based on the occurrence of SLE according to the ACR criteria associated with clinical picture, histopathological findings, direct immunofluorescence tests and/or other immunologic tests. Lupus nephritis is a serious complication occurring in 50% of BSLE cases with a variable manifestation. By this case of BSLE with nephrotic syndrome, we highlight the clinical presentation, histologic and immunologic findings in the diagnosis of BSLE. The diagnosis of BSLE should be considered in cases of blistering eruption suspicious for autoimmune disease, and a routinely systemic examination is essential for screening for organ involvements.