
1. INTRODUCTION
Hepatitis E virus (HEV) is a non-enveloped, positive-stranded RNA virus with a length of approximately 7.2 kb, comprising of 3 open reading frames (ORF 1-3) [1]. Globally, an estimated 20 million HEV infections are reported annually, leading to an estimated 3.3 million symptomatic cases of hepatitis E [2]. The HEV is a member of the Hepeviridae family, which includes the two genera Piscihepevirus and Orthohepevirus [2]. Orthohepeviruses include the four different Orthohepevirus species (A, B, C, D), which may infect a variety of mammalian and avian species. The most common species is orthohepevirus A, which has been isolated from humans, pigs, wild boars, deer, rabbits, and camels [3]. Transmission of orthohepevirus A primarily occurs through the fecal-oral route, often facilitated by fecal contamination of drinking water [4]. This species is characterized by eight HEV genotypes (HEV-1 to HEV-8) distinguished by phylogenetic analysis of complete viral genomes [5],[6]. While HEV-1 and -2 exclusively infect human, causing waterborne epidemics in underdeveloped nations and potentially severe disease in pregnant women [7], HEV-3 and -4 are commonly found in animals like deer, pigs, wild boars and rabbits, with transmission to humans occurring through the consumption of raw or undercooked meat, animal milk, and direct contact with animal feces. HEV-5 and -6 genotypes were recovered from wild boars in Japan [4],[8]–[10], HEV-7, and HEV-8, were recently isolated from camels [11],[12]. HEV-1 and HEV-2 typically cause severe acute hepatitis but not chronic infection [12], but HEV-3, -4, and -7 genotypes may induce both acute and chronic hepatitis in immunocompromised persons [12]–[15]. Human infection genotypes include 1, 2, 3, 4, and 7 [16].
In Vietnam, the most studied diseases include hepatitis A virus (HAV), hepatitis B virus (HBV), and hepatitis C virus (HCV) and some hepatitis D virus (HDV) associated HBV [17],[18] however researching on HEV is still lacking. Detection of HEV can be achieved directly by identifying the presence of its RNA using nucleic acid amplification testing (NAT) or capsid antigens or indirect via assessing the anti-HEV antibodies using immunological techniques [19]. In NATs, popular techniques used for HEV detection are reverse transcriptase polymerase chain reaction (RT-PCR), real-time PCR (qPCR), and loop-mediated isothermal amplification assay (LAMP) [19]. Additionally, nested PCR, which is cost-effective albeit less sensitive, is employed for HEV RNA detection. Most current NATs for HEV primarily target ORF-3, a conserved region, but more geared towards bloodborne HEV [19].
In this study, a nested PCR protocol targeting ORF-1 was specifically developed for the detection of HEV RNA in farmed wild boar samples. It was to provide an alternative method for HEV detection, particularly for animal samples, which is useful for epidemiological research and ultimately enables more reliable and precise diagnosis of HEV in both research and clinical settings.
2. MATERIALS AND METHODS
Samples of farmed wild boar feces were meticulously collected under aseptic conditions. Specifically, a minimum of 1 gram of feces was gathered using disposable sterilized collection tools and placed into a sterile plastic container. Subsequently, the collected fecal material was then suspended in 1 milliliter of a viral transport medium using Nucleic Acid Preservation (NAP) Buffer [20]. When collecting farmed wild boar rectal swabs samples, disposable sterilized swabs were carefully inserted into the rectal area, rotated to collect the specimen, and then immediately placed into sterile transport containers with no transport medium.
Furthermore, liver tissue samples were collected from the market in Ho Chi Minh City, Vietnam. The domesticated pigs ranged in age from 6 weeks to 4 years as determined by the seller in the market, however, the specific age of the individual sampling pig was not documented. The liver sample was collected using disposable gloves, sterile scalpel blades and scalpel, then immediately placed into a sterile container. All the collected samples were maintained at 4℃ (max. 24 hours), subsequently transported to the laboratory, and frozen at −80℃ until processing.
The study was reviewed in August 2022 and approved by the Animal Ethical and Experimental Committee of the International University - Vietnam National University HCM City in December 2022.
Rectal swabs and fecal suspension in Phosphate-buffered saline (PBS) with ratio 0.1 g rectal swabs in 1 mL PBS were used for RNA extraction. The applicable volume of rectal swabs and the feces suspension was centrifuged at 10,000×g for 2 minutes to remove solids from the sample, while 0.1 mg liver tissues were homogenized with a sterilized mortar and pestle with 1,000 µL PBS. Then, using the QIAamp Viral RNA Mini Kit (Qiagen; Catalog #52906, Lot #166024216, Hilden, Germany), process 140 μL of supernatant from 3 kinds of samples according to the manufacturer’s instructions [21]. Complementary DNA (cDNA) synthesis was performed using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA, USA).
The primers were designed based on the RNA-dependent RNA polymerase region, which belongs to the viral ORF1. Outer primer pairs were HEV-38 (sense) 5’-GAG GCY ATG GTS GAG AAR G-3’ and HEV-39 (antisense) 5’-GCC ATG TTC CAG ACR GTR TTC C-3’; the inner primers were HEV-37 (sense) 5’-GGT TCC GYG CTA TTG ARA ARG-3’ and HEV-27 (antisense) 5’-TCR CCA GAG TGY TTC TTC C-3’. The primers were purchased from Eurofins Genomics (Ebersberg, Germany).
F02 and L1 served as replicate samples in our study in which F02 was a positive sample and L1 is a negative fecal sample provided by Vietnamese-German Center for Medical Research. A commercial HEV standard was used as positive control and distilled water, as standard negative control.
Four different conditions, namely protocol 1–4 (Table 1), were utilized in conjunction with 2 nested-PCR programs, A and B (Table 2). The utilization of four distinct protocols and two specific programs plays a pivotal role in optimizing the Nested PCR process. These protocols involved variations in concentrations and components, such as Buffer, Polymerase, dNTPs, MgCl2, as well as primers and templates.
The amplification products (306 bp) were visualized by electrophoresis on 1.7% agarose gels in Tris-acetate-EDTA buffer (TAE). Five μL of each PCR product were mixed with 1 μL loading dye and loaded on the gel. Electrophoresis was performed at a constant 110 V until the bromophenol blue traveled 75% of the gel length. The gel for was incubated approximately 15 mins in TAE containing ethidium bromide and visualized on Life Technologies E-Gel Imager (Thermo Fisher Scientific, Waltham, MA, USA).
3. RESULTS
A total of 133 samples were collected from 2 wild boar farms and markets in Ho Chi Minh City. Among those, 25 rectal swabs and 5 fecal samples were from Farm 1, located in Cu Chi; 35 rectal swabs and 20 fecal samples were from Farm 2, situated in District 9 (Supplementary Table S1, Supplementary Table S2) and 48 liver tissue of domesticated pigs were collected from several markets in Ho Chi Minh City (Supplementary Table S3).
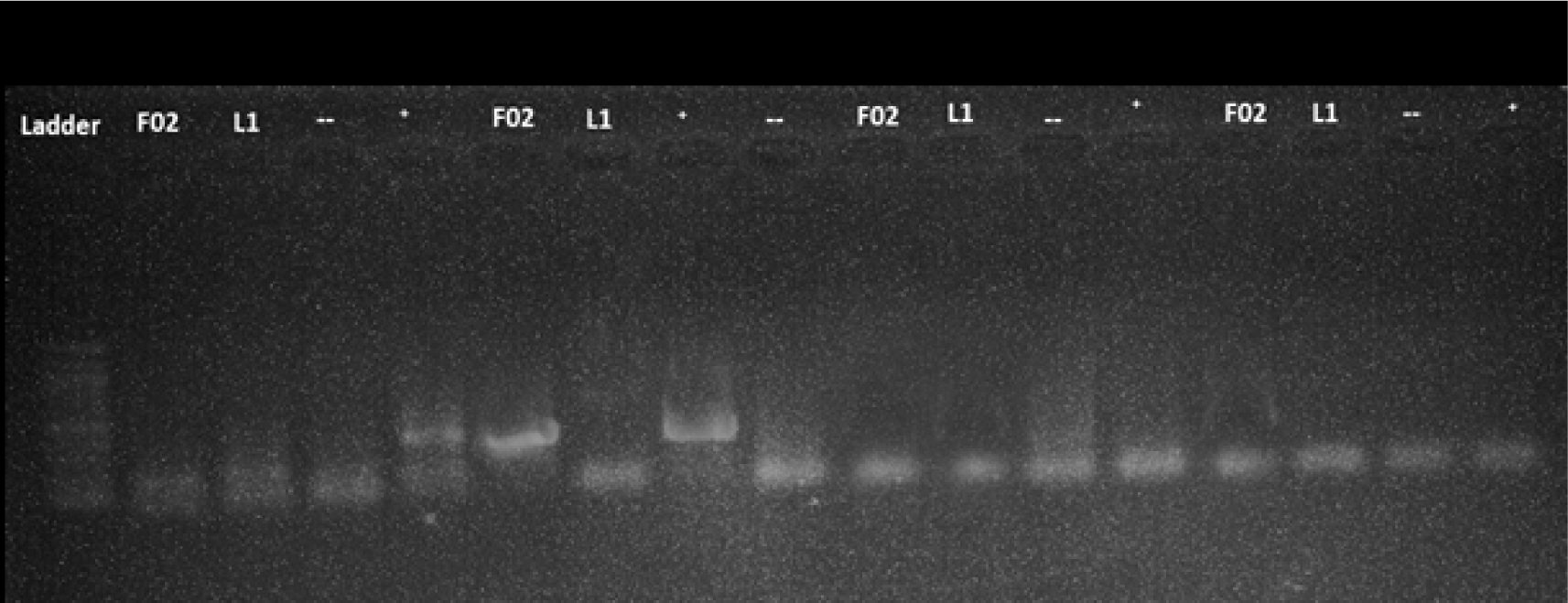
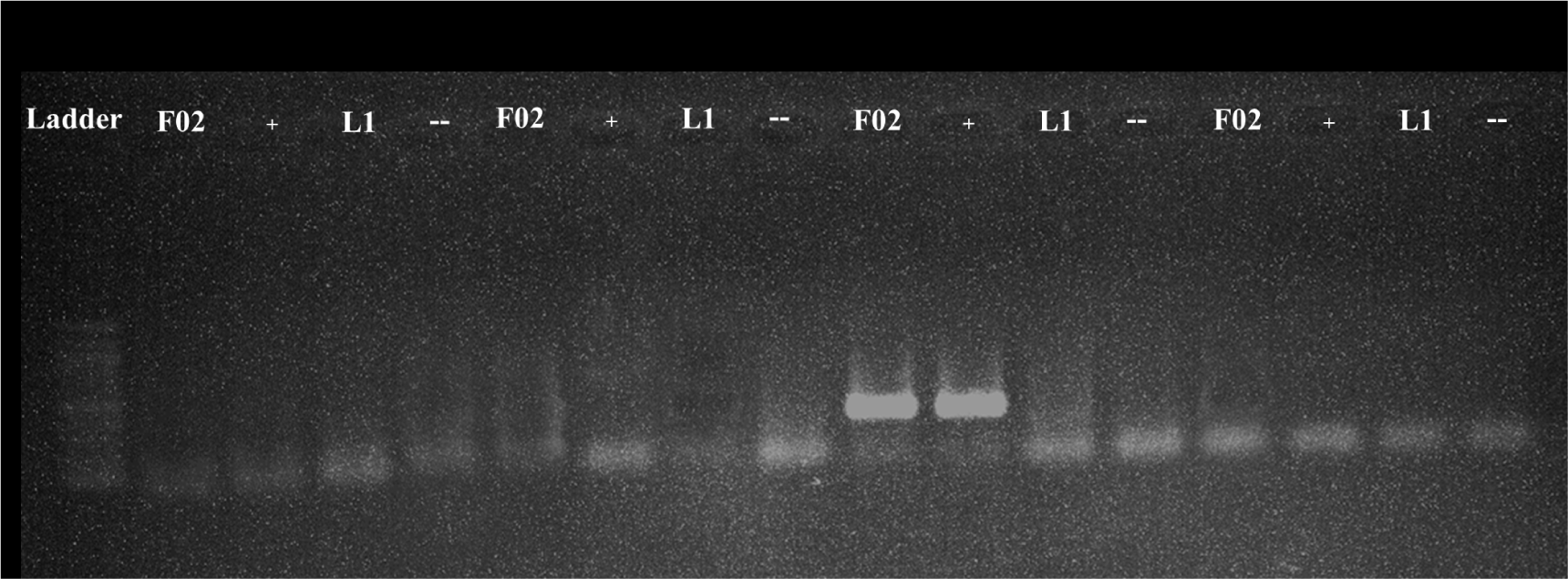
All the samples were tested using protocol 1 and 2 with program A and B. However, the detection was failed as no amplified product was seen including the positive control (F02) and standard positive control (Table 3). The representative results of protocol 1 and 2 with positive and negative standards were shown for program A (Fig. 1) and program B (Fig. 2).
| Protocol | Rectal swabs, faces samples, liver tissues (n=133) | |
|---|---|---|
| Program A | Program B | |
| Protocol 1 | 0 | 0 |
| Protocol 2 | 0 | 0 |
| Protocol 3 | 28 | 0 |
| Protocol 4 | 0 | 28 |
In contrast to protocol 1 and 2, protocol 3 and 4 exhibited a successful amplification of the target. Notably, protocol 3 was effective when paired with program A, while protocol 4 yielded results when paired with program B. The representative results of protocol 3 and 4 with positive and negative controls were shown for program A (Fig. 1) and program B (Fig. 2).
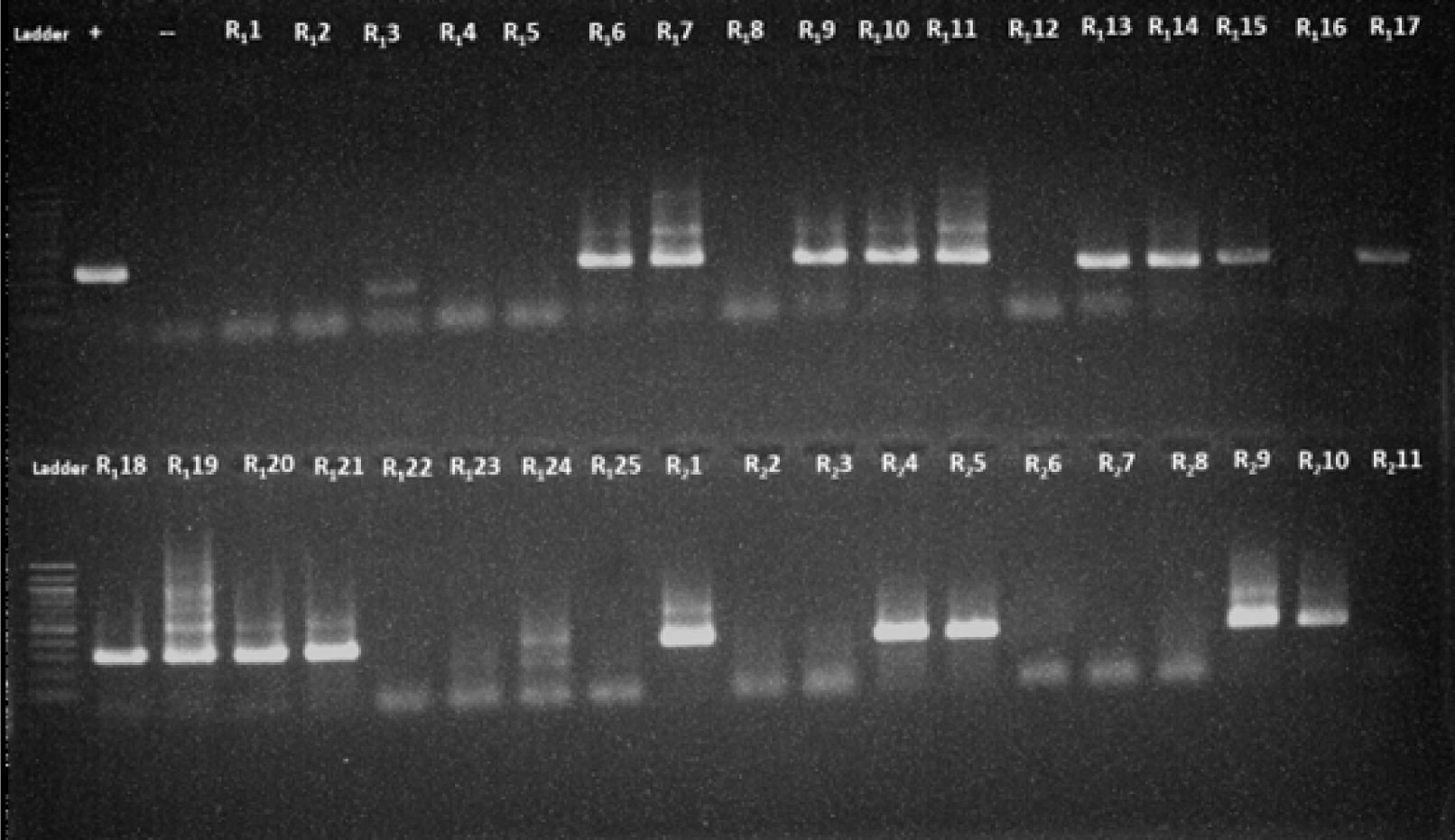
With the combination of protocol 4 and program B, out of the 133 samples, 28 samples (21%) tested positive for amplified products (Table 3, Fig. 3). These findings indicated that protocol 3 and 4 were more effective in detecting the target compared to protocol 1 and 2.
When protocol 3 was combined with program A, a total of 28 samples (21%) tested positive for amplified products. However, the bands observed in these samples were of low intensity and appeared faint, indicating a potential limitation in the detection process (Fig. 1). On the other hand, the combination of protocol 3 with program B resulted in a 0% detection rate, suggesting that program B was not effective in amplifying the target sequence with protocol 3 (Table 3, Fig. 2).
When protocol 4 was paired with program A, no amplified products were detected (Fig. 1). However, when protocol 4 was combined with program B, the amplified products were found in 28 samples (21%) (Table 3), with clearer and higher- intensity specific bands, indicating a higher level of accuracy and specificity (Figs. 2 and 3). All the PCR images with positive results were included in supplementary figures (Supplementary Figs. S1 and S2).
4. DISCUSSION
A total of 133 samples collected from two farms and a market in Ho Chi Minh City, Vietnam were analyzed using different PCR protocols and programs. Initial testing with protocol 1 and 2 did not yield any positive results, resulting in a 0% detection rate for these protocols. However, protocol 3 and 4 showed more success, reaching 21% of the samples testing positive for the amplified product. When protocol 3 was combined with program A, 21% of the samples tested positive, but with low-intensity bands. In contrast, protocol 3 combined with program B resulted in a 0% detection rate. Protocol 4 combined with program A also yielded a 0% detection rate, while protocol 4 with program B showed clearer and more specific bands, indicating improved accuracy and specificity.
The concentrations of MgCl2 and primers play the most crucial roles in the PCR assay [22]. MgCl2 is an essential cofactor for DNA polymerase activity, including the Taq polymerase commonly used in PCR. It helps stabilize the DNA double helix, promotes primer annealing to the template DNA, and facilitates the enzymatic replication process. Similarly, the primer concentration is critical for successful amplification. Primers are short DNA sequences that flank the target region and provide a starting point for DNA synthesis. Their concentration affects the efficiency and specificity of primer annealing to the template DNA during PCR. Optimizing the concentrations of MgCl2 and primers is essential for maximizing the sensitivity, specificity, and efficiency of the nested PCR assay. It involves empirical testing and careful adjustment to achieve the best amplification conditions for the target DNA sequence while minimizing non-specific amplification and other artifacts.
In protocol 1, 2, and 4, the concentrations of MgCl2 and primers were optimized to enhance the detection of HEV using the nested PCR method. For MgCl2, protocol 4 utilized a suitable concentration of 25 mmol (0.3 μL in the outer stage and 0.25 μL in the inner stage). This concentration ensures the availability of divalent cations required for the efficient enzymatic activity of the DNA polymerase. In terms of primers, protocol 4 used a higher concentration of HEV38/HEV39 (0.6 μL in the outer stage) and HEV37/HEV27 (0.9 μL in the inner stage) compared to protocol 1 and 2 (0.5 μL in both outer and inner stage). This higher primer concentration can enhance the amplification of the target HEV sequences and improve the sensitivity of the PCR assay.
In protocol 3, the optimized combination of 5X Q5 Reaction Buffer and Q5 Hot Start DNA Polymerase (New England Biolabs, Inc) was successfully utilized. Q5 Hot Start DNA Polymerase is a high-fidelity DNA polymerase that offers thermostability and a hot start feature. It possesses 3’→5’ exonuclease activity and is fused with a processivity-enhancing Sso7d domain to support robust DNA amplification. This polymerase is specifically designed for high-fidelity amplification, with an error rate approximately 280 times lower than that of Taq DNA Polymerase. It is particularly suitable for cloning purposes and can be employed for amplifying long or challenging amplicons. The 5X Q5 Reaction Buffer contains 2 mM Mg2+, which is supplied with the Q5 Hot Start DNA Polymerase to become an optimized buffer system. This buffer system is particularly suitable for detecting HEV because the coding sequences of all HEV samples analyzed showed an overall GC content value exceeding 50% [23].
The master mix prepared in protocol 4, after making appropriate adjustments, has shown excellent performance in comparison to protocol 3, which utilized a commercial master mix. This highlights the significance of protocol 4, underscoring the importance of meticulous optimization and customization. The precisely tailored master mix in protocol 4 has exhibited superior efficacy, providing a reliable and effective solution for the nested PCR assay.
The main differences between PCR program A and B are the annealing temperature values and the number of thermal cycles. Optimal results were obtained with an annealing temperature of 56℃ for the primer set in the outer round and an annealing temperature of 54℃ for the primer sets in the inner round. The 2℃ difference in annealing temperatures between the first and second rounds was established to increase the sensitivity and specificity of the nested reactions while also eliminating the possibility of monomer and dimer formation. These protocols were tested on a large number of samples, as depicted in Fig. 3.
The PCR reaction mixture undergoes rapid heating and cooling cycles, enabling strand separation in duplex DNA, annealing of primers to the plus and minus strands of the DNA template, and elongation of the PCR product. Depending on the ideal temperature for DNA polymerase activity and the G-C concentration of the template DNA, the formula begins with a denaturation phase at 95℃. It’s essential to be cautious about the number of cycles employed, as an excessive number of rounds can often lead to the amplification of unwanted secondary products. As evidenced by the results, it was found that utilizing 30 cycles for the outer phase and 36 cycles for the inner phase would yield a significant quantity of PCR products.
The diagnosis of HEV infection can be performed via two main methods: serological tests and NAT. Serological tests, such as enzyme immunoassays, are commonly used for detecting HEV infection. Indirect EIA assays are employed to detect immunoglobulin M (IgM) and immunoglobulin G (IgG) anti-HEV antibodies. IgM antibodies become detectable approximately four weeks after infection and can persist for up to six months. IgG antibodies, on the other hand, appear early after infection, often in conjunction with IgM antibodies, and can remain present for several years. The widely used enzyme-linked immunosorbent assay is routinely utilized to identify IgM and IgG anti-HEV antibodies [24]–[26]. An alternative serological test known as the rapid immunochromatographic assay is also available for detecting IgM anti-HEV antibodies. These tests offer a simpler and more cost-effective alternative to traditional EIA assays. Apart from antibody detection, serological assays can also target viral antigens. Viral antigens are present during the early phase of acute HEV infection [24],[25],[19]. This direct approach involves detecting specific proteins, such as capsid protein (ORF2) or phosphoprotein (ORF3), which are indicative of acute infection. When compared to NAT, serological assays are generally simpler and less expensive. They can detect viral antigens even before the onset of symptoms. However, it is important to note that while serological assays tend to be sensitive, their specificity may be relatively lower.
HEV RNA can be found in the blood and fecal three weeks after infection, right before the onset of symptoms. The PCR- based NAT and its variants is considered the gold standard for diagnosing infection. There are several NAT methods often use for detecting HEV, such as RT-PCR, multiplex PCR, qPCR, nested PCR, digital droplet PCR, and LAMP which offer higher specificity and sensitivity compared to EIA. However, they come with certain drawbacks such as high costs, specialized equipment, and expert personnel [16],[27],[28]. Among these methods, qPCR stands out as a highly specific and sensitive technique, widely recognized as the best approach for HEV diagnosis. It operates on the same principles as conventional PCR but with enhanced accuracy. However, in scenarios where sequencing is required for additional purposes such as genotyping and phylogenetic analysis, nested PCR emerges as the optimal choice. The sequencing of the HEV genome involves reverse transcription of HEV RNA, followed by nested PCR amplification of a specific region, and subsequent Sanger sequencing. Typically, the regions targeted for sequencing are ORF1 and ORF2 of the HEV genome [24],[29].
Employing nested PCR followed by sequencing enables researchers to gather crucial insights into the genotype and phylogenetic characteristics of HEV strains. This methodology facilitates a thorough exploration of the virus and its genetic variations, offering a comprehensive understanding. Consequently, nested PCR plays a pivotal role in HEV research, enabling detailed analyses that surpass the capabilities of routine diagnostic techniques.
5. CONCLUSION
HEV is a disease that has received limited research attention, and the absence of standardized diagnostic tests has probably led to an underestimation of the actual number of individuals infected with the virus. Commercial meat products with infectious HEV have been detected in local supermarkets in numerous countries, raising concerns about zoonotic transmission via consumption of undercooked meat. In this study, by combining 4 protocols with 2 PCR programs, the standard nested PCR protocol for HEV detection was successfully optimized. Specifically, the combination of protocol 4 and PCR program B resulted in the generation of high-quality single bands. This optimized condition demonstrates improved sensitivity and specificity for HEV detection. In conclusion, this novel condition presents a promising avenue for the sensitive detection of HEV, providing a reliable and suitable product that facilitates accurate sequencing.
SUPPLEMENTARY MATERIALS
Supplementary materials are only available online from: https://doi.org/10.32895/UMP.MPR.8.2.11