
1. INTRODUCTION
Asthma is characterized by chronic inflammation and structural alterations in the airways, impacting an estimated 9.79% of people across various age groups [1,2]. The World Health Organization has highlighted that asthma leads to 455,000 fatalities in 2019, with a disproportionate impact on low-income countries [3]. Notably, the prevalence of asthma has been increasing annually, contributing significantly to the global economic burden. Between 1990 and 2019, the global number of asthmatic cases gradually increased from 6,487,957.18 to 7,604,488.39 [4,5].
Furthermore, the economic impact of asthma is significant worldwide [4,5]. Despite this, traditional anti-asthmatic medications failed for 5% of patients, while 1%–4% continue to experience severe or uncontrolled asthma symptoms [6,7]. This underscores the challenges faced by healthcare systems in effectively managing asthma, which may be attributed to the lack of specialized biomarkers for various asthma phenotypes and limited biologic resources.
Research into the role of neutrophils in asthma exacerbations and targeted treatment modalities is limited due to the lack of well-defined clinical diagnosis criteria. Based on sputum profiles, four unique asthma endotypes have been identified: eosinophilic asthma (EA), neutrophilic dominant asthma, mixed-granulocytic asthma (MGA), and pauci-granulocytic asthma. EA characterized by sputum eosinophilia without sputum neutrophilia, is more common and has been extensively studied. This has led to a better understanding of its mechanisms and the development of specific biomarkers and treatments. Neutrophilic dominant asthma, on the other hand, is identified by an increase in neutrophils without a rise in eosinophils, while mixed granulocytic asthma involves elevations in both eosinophils and neutrophils. Most studies use sputum neutrophilia (with or without sputum eosinophilia) to recognize neutrophilic asthma (NA) [8]. NA is frequently associated with a reduced response to conventional anti-inflammatory treatments such as corticosteroids and type 2 biologics [9-11]. NA is typically an adult-onset phenotype and may be associated with obesity, smoking, occupational exposure, respiratory infections, and air pollution [9-11], which are significant global burdens.
Treatment options for neutrophilic inflammation remain scarce and variable. For instance, azithromycin therapy has shown promise in reducing asthma exacerbations in patients with NA, suggesting its potential as a supportive treatment [12]. Based on the sputum profiles, interleukin (IL)-8, tumor necrosis factor-alpha (TNF-α), and extracellular DNA (eDNA) are widely accepted as the established biomarkers for neutrophil extracellular traps (NET) formation in severe asthma (SA) [13-15]. However, clinical data on biologics targeting IL-8 and TNF-α for controlling NA show variable results, from no change to slight improvement as reviewed [8]. While preclinical studies in asthma animal models have shown that targeting eDNA can reduce neutrophilia and proinflammatory cytokines, clinical trials have not been extensively established [16]. This highlights a critical need for more comprehensive research and clinical documentation to advance the understanding and management of this challenging asthma endotype.
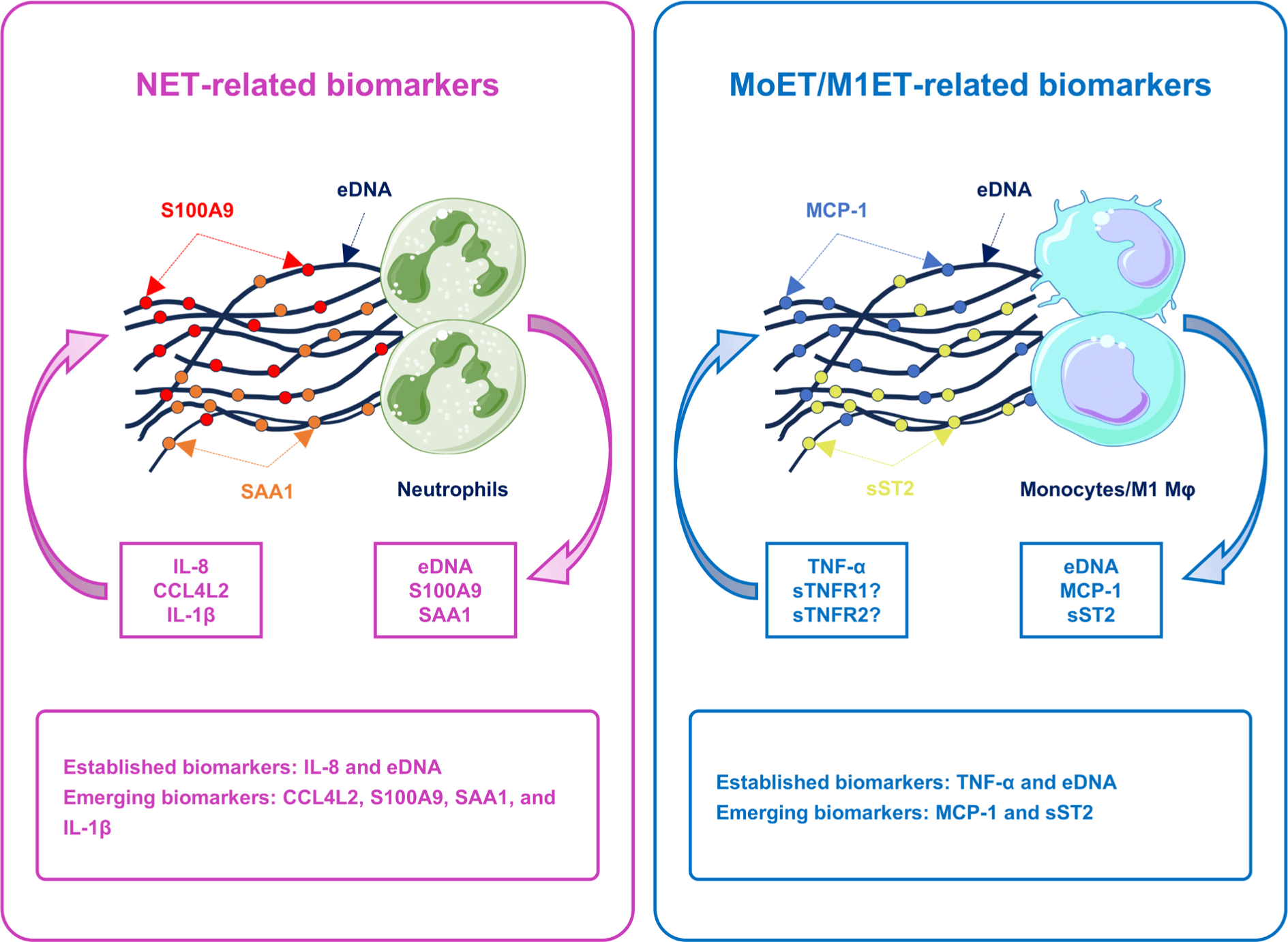
Effective management of NA necessitates a thorough understanding of its fundamental mechanisms and biomarkers. Monocytes, M1 macrophages (Mφs), and neutrophils could exert their effects by discharging web-like structures made of chromatin fibers, or eDNA traps [17,18]. These formations, including NETs, monocyte extracellular traps (MoETs), and M1Mφs extracellular traps (M1ETs), function as blockades that capture and neutralize pathogens, thereby preventing their spread and ultimately destroying them via the high levels of granule proteins and reactive oxygen species (ROS) [17,18]. In NA, an excessive production of these eDNA traps promotes airway inflammation and damages the airway epithelial cells (AECs), thereby diminishing the efficacy of standard asthma treatments [17-19]. Recently, emerging biomarkers for NA have been categorized into: (1) NET-related biomarkers, including C-C motif chemokine ligand 4 like 2 (CCL4L2), serum amyloid A1 (SAA1) [20], calcium-binding protein A9 (S100A9) [21], and ovarian tumor protease deubiquitinase with linear linkage specificity (otulin)/IL-1β [22]; (2) MoET/M1ET-related biomarkers, such as soluble suppression of tumorigenicity 2 (sST2) [23], and monocyte chemoattractant protein-1 (MCP-1) [17] (Fig. 1). This review presents an exhaustive overview of NA, covering its pathophysiology, diagnostic criteria, and prospective research on biomarkers.
2. THE DIAGNOSIS AND CLASSIFICATION OF ASTHMA PHENOTYPES/ENDOTYPES
Diagnosing asthma requires a comprehensive evaluation of key components such as airway inflammation, hyperresponsiveness, bronchial obstruction, and associated symptoms. Nevertheless, in clinical practice, asthma diagnosis is often oversimplified, primarily relying on medical history and symptoms such as wheezing, coughing, breathlessness, and chest tightness, as well as trials of short-acting bronchodilators [24,25]. However, diagnosing asthma solely based on clinical examination has an accuracy of only 63% to 74% and shows a poor correlation with airway obstruction [24,25]. This underscores the necessity of spirometry tests for confirming the diagnosis and assessing asthma severity [24,25]. Additionally, the bronchodilator reversibility test, which is based on the relative change in FEV1 value between the baseline and bronchodilator-taking, was used to evaluate airway hyperresponsiveness [26].
The clinical outcomes of asthma vary due to phenotypic heterogeneity and genetic predispositions, emphasizing the importance of asthma endotyping/phenotyping in effective asthma management [9-11]. Phenotypes refer to the observable characteristics of asthma, such as clinical symptoms (cough), comorbidities (other allergic diseases, obesity, and cardiovascular disease), the onset of disease, environmental exposures (such as occupational agents, cigarette smoke, air pollution, and cold dry air), and genetic factors [27,28]. The literature identifies several phenotypes, such as “non-atopic” or “intrinsic” versus “atopic” or “extrinsic”, infectious asthma, and aspirin-exacerbated respiratory disease [27-29]. Despite sharing similar clinical symptoms, patients may exhibit diverse responses to the same therapeutic interventions. Hence, the current approach aims to link distinct pathophysiological mechanisms at a cellular and molecular level to phenotypes, known as endotyping. Notably, the differentiation between type 2-high and type 2-low asthma endotypes based on non-invasive tests (e.g., fractional exhaled nitric oxide, blood/sputum eosinophils, serum total immunoglobulin E, and serum periostin levels) has garnered attention [8,30-32]. While type 2-high asthma is characterized by elevated levels of type 2 inflammatory biomarkers and eosinophil activation, type 2-low asthma is commonly observed in non-allergic and non-EA patients.
Despite treatment with type 2 biologics, such as omalizumab, some patients with type 2-high asthma still face significant clinical challenges due to persistent airway inflammation and obstruction. This subgroup of patients experiences frequent hospitalizations, severe uncontrolled symptoms, airflow limitation, and the presence of neutrophilia in their airways [33]. Additionally, the increase in sputum neutrophil counts has been reported to correlate with a decrease in lung functional parameters, including pre- and post-FEV1, and the FEV1/FVC ratio [21,34,35]. Therefore, these events highlight a necessity to redefine asthma endotypes [31,36,37]. Recently, researchers suggested asthma classification strategies based on sputum profiles and have identified four different categories: EA, neutrophilic dominant asthma, NA, MGA, and pauci-granulocytic asthma. Of these, NA and MGA have emerged as subtypes with airway inflammation dominated by neutrophils, either alone or in combination with eosinophils [1]. It is acknowledged that the threshold for sputum neutrophilia varies. Several studies have found thresholds for abnormal neutrophil counts or neutrophilia in sputum from healthy controls (HCs), ranging between 49% and 93%; among them, most authors use a threshold between 61% and 76% to investigate the mechanistic pathways in NA [38-40]. In the line of sputum analysis, the characteristics of NA, including more severe phenotype, high-dose inhaled corticosteroid (ICS) requirement, and lower lung functions, were confirmed based on lung biopsies. There is a report that classified NA vs non-NA groups based on bronchial lamina propria neutrophil content (≥47.17 vs <47.17 neutrophils·mm−2) [41]. Altogether, the lack of consensus on diagnostic criteria poses challenges for the diagnosis and treatment of NA.
3. FUNCTION OF IMMUNE CELLS IN neutrophilic asthma
Neutrophils, the most abundant type of circulating leukocytes, play a crucial role in pathogen elimination (including bacteria and fungi) from the body [42]. These specialized cells respond to inflammatory signals and migrate to sites of infection or tissue damage through a process called chemotaxis, which allows them to move toward the source of the signal [43]. This is facilitated by the release of chemokines, such as IL-8, which also attracts other neutrophils to the site. Additionally, they also release cytokines, such as TNF-α, which helps recruit other immune cells to the inflammatory site. To kill pathogens and break down damaged tissue, neutrophils release enzymes, ROS, and NETs [44]. The activation of neutrophils is modulated by various stimuli, including bacterial products (e.g., lipopolysaccharides [LPS]), inflammatory mediators from Mφs (IL-6, IL-1β, and TNF-α), and lymphocytes (such as interferon-gamma [IFN-γ], IL-17, and IL-22) [21,22]. However, in the context of certain conditions (referred to as “NA”), neutrophils are abnormally recruited into the airways and turn to a more activated state [21,45]. Consequently, various proinflammatory cytokines, chemokines, proteases, and other enzymes are released, causing damage to the airway epithelium, and potentially leading to airway remodelling.
Neutrophils could release NETs, which have a complex three-dimensional structure and are the primary mechanism by which neutrophil activation causes airway inflammation in SA [46,47]. NETs caused damage to AECs and produced autoantigens and proinflammatory cytokines. Besides, myeloperoxidase (MPO), for example, worked in combination with neutrophil elastase (NE) to stimulate chromatin decondensation, which was a key step in the creation of NETs [48]. Furthermore, NE worked with cathepsin G to produce active cleavage products of full-length IL-33, which played an important role in eosinophilic and neutrophilic inflammation in SA [23,49-52]. NE also affected the production of inflammatory cytokines and chemokines (e.g., IL-4, IL-5, and IL-13), as well as genes associated with airway smooth muscle cell proliferation and extracellular matrix deposition in asthma [53]. Furthermore, NETs could induce eosinophil activation to produce eosinophil-related cytokines. Thus, NETs played an important role in exacerbating both neutrophilic and eosinophilic airway inflammation in patients with SA [54,55].
Unlike neutrophils, which are the first cells to arrive at the site of inflammation (typically within 1-2 hours after the onset of inflammation), monocytes are recruited to the site of inflammation later than neutrophils, typically within 2–4 hours after the onset of inflammation [56]. In the lungs, monocytes could differentiate into Mφs which play a critical role in both innate and adaptive immunity by engulfing and destroying pathogens, clearing cellular debris, and promoting wound healing [57-59]. Although the abundance of Mφs remains a subject of debate—some studies report an increase, while others suggest no change or even a decrease- there was consensus among researchers regarding Mφ dysfunction in asthma [60,61].
The impact of Mφ dysfunction extends to asthma phenotypes, specifically distinguishing EA and NA. In response to the tissue microenvironment or interactions with different subsets of innate lymphoid cells (ILCs), Mφs could differentiate into two distinct phenotypes [62]. Mφs could differentiate into the classically activated M1Mφ phenotype upon the stimulation of LPS, IFN-γ, and granulocyte-Mφ colony-stimulating factor, releasing proinflammatory cytokines, including TNF-α, IL-6, and IL-1β. In contrast, the alternative activation of the M2Mφ phenotype is induced by IL-4, IL-13, and transforming growth factor beta (TGF-β), which then releases TGF-β1 and vascular endothelial growth factor [17,21,57-59]. While M2Mφs play a key role in promoting airway remodeling in EA, M1Mφs contribute to airway inflammation and are often associated with neutrophilic airway inflammation in SA [17,21,63]. Additionally, M1Mφs released M1ETs, which induced damage to AECs, recruited neutrophils, and activated inflammatory responses [17]. This activation could lead to increased migration of inflammatory cells and enhanced production of proinflammatory cytokines, contributing to the overall inflammatory milieu in asthmatic airways.
Lymphocytes play a crucial role in the immune system. They are responsible for the direct cell-mediated killing of infected or neoplastic cells, regulating the interaction and coordination of the immune cells, and developing immunological memory [64,65]. They are attracted to the site by chemokines and other inflammatory mediators [66]. Dysfunction of lymphocytes can lead to various diseases, including autoimmune disorders, immunodeficiency disorders, and even asthma [44]. Different subsets of CD4+ T cells—such as Th1, Th2, and Th17—play distinct roles in inflammation by secreting their specific cytokines [44]. For example, Th1 cells produce IFN-γ, TNF-α, and IL-2, promoting chronic inflammation and autoimmunity in SA. Th2 cells produce IL-4, IL-5, and IL-13, which aid in eosinophil recruitment, activation, and airway remodeling. Th17 cells contribute to chronic neutrophilic inflammation by producing pro-inflammatory cytokines like IL-17, IL-22, and IL-23 [8,67]. Additionally, ILCs including ILC1s, ILC2s, and ILC3s—which mirror the cytokine profiles of Th1, Th2, and Th17 cells, respectively, could contribute to inflammatory responses in the asthmatic airways [68]. ILC3s released IL-8 which could recruit neutrophils into the airways and promote neutrophilic airway inflammation in SA. Importantly, these events were not controlled by steroid treatment [69]. Therefore, by considering the intricate balance between these immune components, regulating these lymphocyte activities could reduce steroid resistance and aid in the development of targeted biologics for asthma treatment.
4. BIOMARKERS OF NEUTROPHILIC ASTHMA
Examining the NET composition and its primary stimulator, IL-8, in clinical specimens from asthmatic patients—such as eDNA, citrullinated histone H3 (CitH3), and NE—could serve as biological indicators of NET. Although asthmatic patients had greater levels of MPO, NE, and CitH3 than HCs, the links between these cytokines and NA were not well-established in the literature [70-72]. Furthermore, classical monocytes were important inflammatory cells that coexisted with neutrophils in the airways of patients with NA [73]. TNF-α and its receptor expressions differed between sputum and blood, suggesting that classical monocytes might be more active in asthma patients [74]. Monocytes could differentiate into alveolar Mφ, which were then popularized into M1Mφ and released proinflammatory cytokines including TNF-α and IL-1β [17]. In return, TNF-α activated monocytes and M1Mφ, making it a reliable biomarker for SA. These findings indicated that the acknowledged biomarkers for NA included increased IL-8, TNF-α, and eDNA levels in sputum (Table 1).
| Biomarkers, reference | Year | Patient recruitment | Clinical results | Key mechanisms |
|---|---|---|---|---|
| Spu IL-8 [15] | 2001 |
EA (Spu Eos≥2.5%) Non-EA (Spu Neu<2.5%) |
Non-EA>EA Correlation of NETs: +Spu Neu |
None |
| Neutrophil autophagy and eDNA [18] | 2016 |
SA1) Non-SA1) |
SA>non-SA Correlation of NETs: +Autophagy –FEV1/FVC |
NETs induced: ↑ Cell death and detachment ↓ Tight-junction proteins in AECs ↑ The release of IL-8 from AECs ↑ The release of ECP and EDN from eosinophils |
| Spu eDNA [13] | 2016 |
NA (Spu Neu>61%) Non-NA (Spu Neu≤61%) |
NA>non-NA Association Correlation: +ACQ +Spu CXCL8 and IL-1β +NLRP3 gene expression –FEV1 –FEV1/FVC |
None |
| Spu eDNA [14] | 2019 | SA |
Association: –ACT +Chronic bronchitis +Use of oral corticosteroids +NE-DNA and CitH3-DNA +Spu IL-1β and caspase-1 activity Correlation: +Spu Neu +Spu MPO |
NETs induced: The release of IL-6, IL-8, and G6PD from AECs |
| Spu TNFR1 and TNFR2 [80] | 2021 |
NA (Spu Neu≥61%, Spu Eos<3%), EA (SPu Neu<61%, Spu Eos≥3%), MGA (Spu Neu≥61%, Spu Eos≥3%), PGA (Spu Neu<61%, Spu Eos<3%) |
NA>EA, NA>PGA Association: +Asthma severity +Asthma exacerbation rates |
None |
Recent research has highlighted that NET formation could also be triggered by S100A9, SAA1, and IL-1β, acting through downstream intracellular mediators, including ROS. These ROS, in turn, activated neutrophils to release MPO and NE, amplifying neutrophilic airway inflammation in SA. Consequently, S100A9, SAA1, and IL-1β could be potential indicators for NA identification and candidates for developing novel therapeutics for SA (Table 2).
| Biomarkers, reference | Year | Patient recruitment | Clinical results | Key mechanisms |
|---|---|---|---|---|
| Spu S100A9 [45] | 2017 |
NA (Spu Neu≥60%, Spu Eos<3%), EA (SPu Neu<60%, Spu Eos≥3%), MGA (Spu Neu≥60%, Spu Eos≥3%), PGA (Spu Neu<60%, Spu Eos<3%) |
NA>EA and NA>PGA Correlation: +Spu Neu |
S100A9 induced: ↑The recruitment of Neu and Mφ ↑The release of IL-1β, IL-17, and IFN-γ |
| Serum S100A9 [21] | 2021 |
NA (Spu Neu≥65%) Non-NA (Spu Neu<65%) |
NA>non-NA Association: +Serum IL-6, IL-17A, and TNF-α +Spu Neu |
S100A9 induced: ↓Tight-junction proteins in AECs ↑The release of IL-8 from AECs ↑The formation of NETs from Neu ↑The release of cytokines from Mφ |
| Serum SAA1 [20] | 2021 |
NA (Spu Neu≥65%) Non-NA (Spu Neu<65%) |
NA>non-NA Association: +Serum S100A9 and IL-6 +Spu Neu |
SAA1 induced: ↑The release of IL-6, IL-1β, and S100A9 from AECs ↑The formation of NETs from Neu ↑The release of cytokines from Mφ ↑The release of IL-17A from CD4+ T cells |
| Plasma CCL4 [16] | 2023 |
ICS non-responders ICS responders |
ICS non-responders>ICS responders Correlation: –FEV1, FEV1/FVC, and PEF25%–75% |
CCL4L2 induced: ↑The formation of NETs from Neu ↑The recruitment of Neu ↑The release of IL-1β, IL-6, and IL-17 |
| Spu IL-1β [22] | 2024 |
NA (Spu Neu≥65%, Spu Eos<3%) EA (Spu Eos≥3%, Spu Neu<65%) |
NA (with high-dose ICSs)>EA Correlation: +Spu MPO and MMP-9 |
Downregulation of otulin induced: ↑The release of IL-1β from Mφ and AECs IL-1β induced: ↑The recruitment of Neu and ILC3s ↑The release of cytokines from Neu and ILC3 |
Similar to NET, monocytes and M1Mφ could release ET, consisting of DNA, CitH3, and various proteins. These structures activated neutrophils by directly stimulating them to induce NET formation and indirectly activating AECs and group 3 innate lymphoid cells (ILC3s) to release cytokines that promoted neutrophil migration and activation. Notably, monocytes and M1Mφ obtained from patients with SA exhibited significantly higher levels of eDNA compared to those from patients with non-SA and HCs [17]. Given the link between serum MCP-1 and sST2 with eDNA from monocytes and M1Mφ, these molecules might be important predictors of MoETs/M1ETs in SA (Table 3).
| Biomarkers, reference | Year | Patient recruitment | Clinical results | Key mechanisms |
|---|---|---|---|---|
| Serum MCP-1 and sST2, and monocytes and M1Mφ eDNA [17] | 2023 |
SA1) Non-SA1) |
SA>non-SA Correlation of MoETs/M1ETs: +Serum MCP-1 and sST2 –FEV1% |
MoETs/M1ETs induced: ↓Tight-junction proteins in AECs 2003;↑The release of cytokines from AECs 2003;↑The formation of NETs from neutrophils 2003;↑ILC3 polarization 2003;↑The release of IL-17A, IL-22, and IFN-γ from ILCs |
| Serum sST2 [23] | 2023 | UA2) PCA2) CA2) | UA>PCA/CA Correlation: –FEV1% Association: +Serum S100A9, MPO, and IL-8 |
IL-33 induced: ↑The formation of NETs from neutrophils ↑The release of IL-6, IFN-γ, and TNF-α from Mφ |
SA and non-SA were defined according to the International European Respiratory Society/American Thoracic Society Guidelines [107].
UA, PCA, and CA were determined following the Global Initiative for Asthma guideline [12].
Here, we presented an overview of established and emerging biomarkers for predicting NET and MoET/M1ET formation in SA.
In recent years, there has been a concerted effort to identify specific molecular targets for diagnosing and managing NA. Research studies have implicated various inflammatory cytokines and chemokines and their downstream signaling pathways associated with neutrophil activation in SA. For instance, our team reported a close association between two promoter single nucleotide polymorphisms in the ATG5 gene (–769T>C and –335G>A) and the ATG7 gene (–100A>G and 25108G>A) and NA characteristics (such as sputum neutrophil counts and serum IL-8 levels) [75]. These correlations suggest that autophagy activation could contribute to the development of NA. Additionally, IL-8 has been shown to induce autophagy signaling pathway activation, leading to the formation of NETs in patients with SA [18]. Elevated levels of IL-8 in the airways have been positively associated with sputum neutrophil counts and asthma severity [15]. These findings have opened new possibilities for developing novel biologic treatments tailored to this subtype. However, clinical trials targeting the blockade of IL-8 yielded disappointing results [8]. The lack of efficacy of these studies may be due to the heterogeneity of these patient subgroups with poorly controlled asthma and these required the development of targeted therapies that specifically inhibit neutrophil activity in the airways.
Wright et al. performed immunofluorescence staining on sputum specimens from patients with NA and observed co-localization of eDNA with NE, suggesting the presence of NETs in asthmatic airways. Moreover, significantly higher eDNA levels, evaluated by Picogreen assay, were noted in patients with SA, and negatively correlated with ACQ scores and FEV1%, indicating the role of NETs in severe uncontrolled asthma [13]. Similarly, findings from the SA Research Program indicated that patients with high sputum eDNA levels had an increased risk of airway neutrophilic inflammation, elevated NE-DNA and CitH3-DNA, and higher caspase 1 activity and IL-1β levels [14]. In line with these findings, our team reported significantly increased eDNA levels in peripheral blood neutrophils from the patients with SA compared to those with non-SA, correlating negatively with FEV1/FEV% [18]. Given these observations, the cytotoxic effects of NETs on AECs could be mitigated by DNase (an endonuclease degrading eDNA) [16]. This underscores the therapeutic potential of DNase for NET-related diseases. It could be recognized by the significantly reduced DNase activity levels in sputum from asthma patients compared to HCs, which correlated with traits of asthma severity [76]. These findings provide valuable insights into the mechanisms linking NETs and NA and may pave a way for more effective treatments.
Activated monocytes and Mφs by LPS released TNF-α, a cytokine from the TNF superfamily [17]. It is an important inflammatory cytokine with a wide range of effects on the immune and physiological systems, including NA. For example, TNF-α signaling dysfunction activated inflammasomes in Mφs, resulting in IL-1β cleavage and increased neutrophilic airway inflammation. This phenomenon has been observed in NA patients with have reduced otulin levels [22,77]. Moreover, TNF-α could trigger the production of MoETs and M1ETs in severe asthmatic monocytes and Mφs [17]. Therefore, blocking TNF-α signaling might potentially treat extracellular trap-mediated disorders, like NA [17]. Emerging evidence suggests that anti-TNF-α antibodies could restore glucocorticoid sensitivity in an asthmatic mouse model with steroid resistance, suggesting that TNF-α could play an important role in steroid resistance in asthma [78]. However, studies on TNF-α levels in sputum have shown inconsistent results, with levels ranging from increased to unaltered [79,80].
Additionally, TNF-α bound to two receptors, TNF receptor (TNFR)1 and TNFR2, which had two forms: soluble TNFs (sTNFs) and transmembrane TNFs. TNF-α converting enzyme activity produced soluble versions of TNFR1 and TNFR2, which arise from alternative splicing or shedding of membrane-bound receptors [81]. Patients have been shown to exhibit significantly different levels of soluble TNFR1 and TNFR2 sputum, but not serum [80]. These sTNFRs could worsen inflammation by increasing neutrophil recruitment and activation through binding to TNFR1 [82]. Azithromycin has been suggested as an alternate strategy for regulating TNF-α signaling pathways, including decreasing soluble ligand and receptor levels; however, further investigation into underlying mechanisms should be considered.
NETs have been linked to steroid resistance in asthma, but the underlying mechanisms remain unclear. Early evidence suggests that the anti-apoptotic nature of neutrophils in response to steroids might contribute to this resistance [83]. The modernist evidence proposed the contribution of CCL4L2 in this process [16]. CCL4, also known as Mφ inflammatory protein-1β, functions as a chemoattractant, binding to receptors on neutrophils and guiding their migration to the airways in asthma patients. This protein could directly activate neutrophils or do so indirectly through inflammatory cytokines like IL-1, IL-6, and TNF-α released by fibroblasts and Mφs [84]. High levels of CCL4 have been identified as a potential biomarker specifically linked to NA, with elevated levels found in asthma patients but not in those without asthma [85]. Recent studies comparing genes associated with ICS nonresponse through various sequencing methods, revealing increased CCL4L2 mRNA expression in ICS non-responders. This expression is positively correlated with neutrophilia in both human and mouse models. Blocking CCL4L2 reduces NETosis and neutrophilic airway inflammation [16]. These findings suggest that further clinical evaluation of CCL4 blockage could enhance our understanding of asthma endotypes and the role of CCL4 in neutrophil recruitment and activation.
S100A9 (also known as MRP-14), a member of the S100 family of calcium-binding proteins, exhibits the characteristic ability to bind calcium ions and undergo conformational changes in response to fluctuations in calcium levels [86]. This protein has garnered attention due to its involvement in various chronic inflammatory diseases, including sepsis [87], arthritis [88], cancer [89], and NA [21,45]. In patients with NA, elevated levels of S100A9 have been observed in both serum and sputum, correlating with neutrophil activation and increased airway inflammatory markers [21,45]. Primarily expressed in neutrophils and Mφ, S100A9 was released upon cellular activation in response to LPS [21,45]. Functionally, S100A9 could bind to toll-like receptor 4, leading to the subsequent production of pro-inflammatory cytokines (such as TNF-α, IL-6, IL-8, and IL-1β), and amplifying neutrophilic airway inflammation in SA [21,45]. Additionally, S100A9 could induce AEC damage through the degradation of tight-junction proteins [21,45]. Furthermore, by regulating the NET formation and neutrophil activation, anti-S100A9 antibodies hold the potential for mitigating inflammatory cytokines in NA mouse models [21,45]. Collectively, anti-S100A9 antibodies have emerged as a promising therapeutic target for treating NA; however, further clinical trials are required.
Another potential biomarker for predicting NET formation is SAA1. SAA1, a major acute-phase protein composed of 104 amino acids, is encoded by the SAA1 gene located on chromosome 11 [90]. Research has shown that serum SAA1 levels were elevated in patients with asthma compared to HCs; among patients with asthma, these levels were even higher in those with NA [20]. In the context of asthma, AECs act as the first line of defense against pathogens; the stimulator polyinosinic: polycytidylic acid, triggered the secretion of SAA1 from AECs. Once released, SAA1 plays a multifaceted role in the pathogenesis of NA: (1) SAA1 activated AECs and Mφ, leading to the release of pro-inflammatory cytokines (such as IL-6, IL-8, and S100A9); (2) SAA1 contributed to neutrophil recruitment within the airways and enhances neutrophil activation to release NETs; and (3) SAA1 has been implicated in inducing IL-17 expression in CD4+ T cells contributing to the Th17-driven inflammation characteristic of NA [20]. Despite these insights, neutralizing antibodies targeting SAA1 effects are not currently available.
IL-1β stands out as a potent pro-inflammatory cytokine central to the innate immune response. It predominantly originates from activated Mφ and monocytes [91]. Its release is strictly regulated by the inflammasome signaling pathways [91]. Notably, analysis of sputum profiles and isolated Mφ from patients with NA revealed excessive expressions of the nucleotide-binding domain, leucine-rich repeat, and pyrin domain–containing protein 3 inflammasome activation and IL-1β [92]. In the context of asthma, IL-1β orchestrated the recruitment and activation of inflammatory cells, including neutrophils, within the airways [22]. Additionally, IL-1β stimulated the release of other pro-inflammatory cytokines—such as IL-6—from AECs [93]. Chronic exposure to IL-1β contributed to structural changes in the airways, leading to airway remodeling and persistent airflow obstruction [94]. Based on findings from human studies, preclinical trials using mouse models of NA treated with anti-IL-1β antibodies have been conducted to evaluate the effects of these antibodies. Mice administered anti-IL-1β antibodies showed reduced inflammatory cell counts and improved airway responsiveness [14,95]. Targeting the otulin/IL-1β pathway is a promising therapeutic strategy for severe NA. Recent research highlights the role of otulin in regulating inflammasome activation and IL-1β production [22]. Otulin primarily cleaves linear (Met1-linked) polyubiquitin chains, which are crucial for the expression of pro-IL-1β and other inflammasome components via nuclear factor-κB and receptor-interacting protein kinase 1 and 3 [77]. Therefore, loss of otulin function in monocytes/Mφ can lead to uncontrolled inflammasome activation and excessive IL-1β production. Enhancing the levels of otulin or developing molecules that mimic its function could potentially reduce the inflammatory burden in patients with severe or uncontrolled asthma.
MCP-1, also known as C-C motif chemokine ligand 2, belongs to the C-C chemokine family—a group of small proteins that regulate cell trafficking and activation [96]. It is predominantly synthesized by various cell types, including endothelial cells, fibroblasts, epithelial cells, smooth muscle cells, monocytes, and Mφ, in response to oxidative stress, cytokines, or growth factors [96]. Previous studies have indicated that MCP-1 levels are markedly elevated in the serum of patients with SA compared to those with non-SA or HCs [17,97]. Additionally, the MCP-1 G/G genotype has been correlated with asthma severity, highlighting the intricate interplay between MCP-1 and SA [98]. Mechanistically, MCP-1 could recruit and activate monocytes and Mφ, promoting airway inflammation. Furthermore, it may contribute to airway remodeling by facilitating fibrosis and smooth muscle cell proliferation, thereby perpetuating the chronic inflammatory state observed in SA [96]. Consequently, MCP-1 is currently under investigation as a potential therapeutic target for SA. Furthermore, eDNA from monocytes and M1Mφ were positively correlated with blood neutrophil counts and serum levels of MCP-1, which in turn were inversely correlated with lung function (FEV1) [17]. Therefore, we suggested that serum MCP-1 could be the surrogate biomarker for these ET formations.
IL-33 is a critical epithelial alarmin that plays a pivotal role in the pathogenesis of asthma, particularly in the context of NET-mediated airway inflammation. Elevated serum levels of sST2 have been observed in patients with asthma, especially during exacerbations [50]. These levels were correlated with disease severity and asthma control status, suggesting that sST2 could serve as a biomarker for monitoring asthma progression and assessing therapeutic responses [23,50]. Additionally, IL-33 could bind to its ST2 receptor (ST2L), a transmembrane receptor expressed on the surface of neutrophils and Mφ [23]. This event initiated a cascade of signaling events leading to NET formation from neutrophils and the production of pro-inflammatory cytokines from Mφ, including TNF-α, IL-6, and IFN-γ, which are central to the neutrophilic response in SA. Nevertheless, the sST2 acted as a decoy receptor for IL-33, binding to the cytokine and preventing its interaction with the ST2L. By sequestering IL-33, sST2 inhibited type 2 response-related downstream signaling pathways associated with inflammation [49]. However, in the context of NA, the IL-33/sST2 complex could enhance IL-33-induced neutrophilic inflammation [51]. Furthermore, IL-33 plays a role as a chemoattractant for the monocytes migrating toward inflammatory areas; serum sST2 positively correlates with the formation of MoETs and M1ETs. Therefore, targeting the ST2 pathway holds promise as a therapeutic strategy for asthma in clinical trials, particularly in non-type 2 asthma [99,100].
One area of interest is the role of airway autoantigens, microbiomes, microRNAs, and oxidative status in shaping asthma phenotypes and endotypes. For instance, NETs had the potential to trigger the release of autoantigens (such as cytokeratin 18, α-enolase, and tissue transglutaminase) from AECs, which could contribute to the production of autoantibodies against these autoantigens [19]. Notably, elevated levels of these autoantibodies have been observed in patients with SA and those with toluene diisocyanate-induced occupational asthma (a condition typically associated with neutrophil activation profiles) [101-103]. Lactobacillus paracasei-derived extracellular vesicles have shown potential therapeutic effects in asthma, with lower levels observed in NA compared to EA and HCs [104]. This finding suggested a diminished protective immune response in NA, highlighting its potential as a biomarker for distinguishing asthma phenotypes. Additionally, hsa-miR-4517, a microRNA upregulated by Micrococcus luteus-derived extracellular vesicles, suppressed IL-1β production from monocytes, thereby reducing neutrophilic inflammation and presenting a novel therapeutic approach for NA [105]. Moreover, elevated 8-Iso-prostaglandin F2α levels found in NA indicated increased oxidative stress, serving as both a biomarker and a target for therapeutic strategies [106]. Altogether, these three molecules represent promising avenues for understanding and potentially managing NA. Further research into these biomarkers in large cohorts may lead to improved diagnostic tools and targeted therapies for patients with NA.
5. CONCLUSION
NA represents a complex and difficult-to-treat phenotype of asthma that demands a comprehensive understanding of its underlying mechanisms and biomarkers. This review has highlighted the crucial role of inflammatory cells and their ET in the pathogenesis of NA, offering potential biomarkers such as SAA1, S100A9, otulin, MCP-1, and sST2 as promising targets for NA diagnosis and treatment. The identification and validation of these biomarkers could pave the way for achieving precision medicine, thereby improving outcomes for patients with NA. Collaborative efforts among researchers, clinicians, and policymakers are essential in addressing the diagnostic challenges and enhancing the overall management of asthma, ultimately reducing its economic and health burden. By advancing our knowledge of NA and implementing evidence-based interventions, we can move towards more effective and personalized care for asthmatics, ensuring a better quality of life for those affected by this chronic condition.