






1. INTRODUCTION
Alzheimer’s disease (AD) is a major cause of dementia in the elderly [1, 2]. This is the ailment of the central nervous system characterized by the injury of the grey matter in the cerebral cortex with a complex neurodegenerative process leading to progressive cognitive decline and memory loss [3, 4]. Nowadays, millions of people worldwide have been affected by the disease and the number of patients is constantly increasing [1]. Therefore, finding out new drugs which can improve this situation is an imperative task.
The etiology of AD is currently not fully known, although factors including the low levels of acetylcholine (ACh), accumulation of abnormal proteins namely β-amyloid (βA) and τ-protein, homeostasis irregularity of biometals, and oxidative stress, are considered to play significant roles in the pathophysiology of AD [3, 5-7]. At the present time, clinical therapy for AD patients is primarily established upon the cholinergic hypothesis which suggests that the decline of ACh level leads to cognitive and memory deficits, and drugs with ability of inhibiting acetylcholinesterase (AChE) would control symptoms of the disease [4, 8-10].
Acetylcholinesterase inhibitors (AChEIs) including rivastigmine, donepezil and galantamine have been recommended in the treatment of AD for a long period and they have created the most developed group of drugs for the disease up to now [4, 10-13]. For the last few years, a vast number of structures have been tested for acetylcholinesterase inhibitory activities. The variety of AChEI groups have provided a plentiful source of materials for in silico studies (QSAR, docking, pharmacophore...). Among these structures, chalcone has been a group with great concern. Chalcone is a sub-group of flavonoid and is the intermediary in the synthesis process of other fiavonoids [14, 15], pyrazolines [16], isoxazoles [17], and quinolinylpyrimidines [18]. There have been a lot of chalcone compounds with a diverse array of reported bioactivities such as antimalarial [19-20], antiprotozoal [21-23], antibacterial [24-27], antifungal [28-30], antiviral [31-33], antioxidant [34-36], antitumoral [37-39], and other characteristics such as antiinflammatory [40-42], analgesic [42-44], antiulcer [45,46], Recent studies on the bioactivities of chalcone compounds have also revealed their abilities in inhibiting enzymes including urease, α-glucosidase [47, 48], lipoxygenase [49, 50], acetylcholinesterase [51-55], mammalian α-amylase [56, 57], xanthine oxidase [58], monoamine oxidase (MAO) [59-61], and β-secretase [62, 63]. In addition, it was reported that chalcone derivatives exhibit high binding affinity to βA aggregates in vitro, and they could serve as a useful mean for in vivo imaging of βA plaques in Alzheimer’s brain [64]. The studies on bioactivities of chalcone derivatives on the function of human brain promise the finding of new drugs for the treatment of many diseases including AD.
Computational approaches have become a critical component of many drug discovery projects, and molecular docking, which was devised during the early 1980s, has been a highly active research area in this respect. This is a method for placing small molecules into the binding site of their macromolecular targets. The molecular docking can be utilised to model the interaction between a small molecule and a protein at the atomic level, which allow to characterize the behavior of small molecules in the binding site of target proteins, as well as to elucidate fundamental biochemical processes or to identify potential bioactive compounds by virtual screening processes [65].
In this study, a total of 20 chalcone compounds were synthesized and studied for their in vitro AChE inhibitory activities. The molecular binding abilities of these compounds with the enzyme were elucidated by docking procedure to provide useful information for the design and synthesis of others with better bioactivities.
2. MATERIALS AND METHODS
All chemicals were obtained from commercial suppliers, and used without further purification. Melting points were determined on open capillary tubes and are uncorrected (using Gallenkamp apparatus). IR spectra were recorded on a Shimadzu FTIR 8201PC instrument. 1H-NMR spectra were recorded on a Bruker (500 MHz) spectrometer. Chemical shifts were reported in parts per million (ppm) downfield relative to tetramethylsilane as an internal standard. Peak splitting patterns were abbreviated as m (multiplet), s (singlet), bs (broad singlet), d (doublet), bd (broad doublet), t (triplet) and dd (doublet of doublets).
Claisen-Schmidt condensation reaction [66] was applied to synthesize chalcones (Fig. 1). The reaction of acetophenone and benzaldehyde derivatives in KOH/ MeOH was followed by an acidification using a solution of concentrated HCl provided S1-S20 with satisfactory yields after re-crystallized from appropriate solvents. The structures and purities of target compounds were confirmed by IR and 1H-NMR spectra.

Acetophenone and benzaldehyde derivatives (with proportion of 1:1) were dissolved in methanol with stirring. Potassium hydroxide was then added. The resulting solution was stirred at room temperature or cooled by ice as needed. The chemical reaction was monitored by thin layer chromatography. Final mixture was cooled and was acidified by a solution of concentrated HCl to pH ≈ 4-6. The resulting yellow solid was filtered, washed with cold water, and recrystallized from appropriate solvents to give the product.
AChE inhibitory activities of chalcones were determined by Ellman’s [67] colourimetric method using purified acetylcolinesterase from electric eel (Type VI) and acetylthiocholine iodide (ATCI) as a substrate and galantamine as a reference. The assay was performed in 96-well microtiter plates in the same condition for both chalcones and control substance. 25 of 100 mM sodium phosphate buffer pH 8, 25 μL of sample and 25 μL acetylcholinesterase solution containing 0.54 U/mL were mixed in each well of the plate and allowed to incubate for 15 min at 25 °C. Subsequently, 25 μL of a solution of ATCI (15 mM, dissolved in water) and 125 μL of 3 mM DTNB (5,5’-dithio-bis-nitro benzoic acid) were added. The absorbance at 405 nm was read during the first 5 min of the reaction. A control reaction which was considered to have 100% activity was carried out using the same volume of methanol/water instead of tested solutions. All samples were assayed in triplicate. Percentages (%) of AChE inhibitions of tested compounds were calculated from the absorbance values as indicated in equation (1):
Where I is the percent inhibition of acetylcholinesterase; A0E is the absorbance value of the control blank sample with enzyme; A0 is the absorbance value of blank sample; Ac is the absorbance value of the tested sample; A0C is the absorbance value of blank test sample.
The content of each sample is indicated in Table 1. Linear recurrent equations indicating the correlation between common logarithm of the concentrations of investigated compounds (μM) and their percentages of AChE inhibition (%) were built, and from which the IC50 values (concentration that inhibits 50% AChE activity) of studied chalcones were then extrapolated.
| Samples | ATCI | DTNB | Buffer | Chalcone | AChE |
|---|---|---|---|---|---|
| Control blank sample with enzyme (A0E) | + | + | + | - | + |
| Blank sample (A0) | + | + | + | - | - |
| Tested sample (AC) | + | + | + | + | + |
| Blank test sample (A0C) | + | + | + | + | - |
The most active compound against AChE was selected for further kinetic investigation. Tested compound was added into the assay solution and pre-incubated with the enzyme at 25 °C for 15 minutes, followed by the addition of substrate (ATCI). Each assay solution contained one of five different concentrations (5, 10, 20, 30, 40 mM) of substrate (ATCI) mixed in the phosphate buffer (pH 8.0), DTNB (3 mM), AChE (0.54 U/mL) and studied compound (30, 40 μM). Kinetic characterization of the hydrolysis of ATCI catalyzed by AChE was done spectrometrically at 405 nm. A parallel control with no inhibitor in the mixture, allowed adjusting activities to be measured at various times.
The Protein Data Bank crystallographic structure of TcAChE(-)-Galantamine complex (pdb 1DX6) [68] was used as receptor model in this study. The 3D structure of the crystallographic complex was rendered by means of BioSolvelT Leadlt [69], The active site was defined as all the important amino acid residues enclosed within a radius sphere of 6.5 Å centered by the bound ligand, galantamine. All unbound water molecules were eliminated and the structures of amino acid residues were checked before reestablishing the active site of the enzyme.
The 2D and 3D chemical structures of 20 chalcones were built using ChemBioOffice 2008 program [70], The structures of molecules were optimized by the energy minimization and molecular dynamic modules in SYBYL-X 2.0 [71]. In the energy minimization process, Conj Grad method was chosen and the structures of molecules were optimized until converged to a minimum energy change of 0.00001 kcal.mol-1. Gasteiger-Huckel charges were assigned to the structure atoms and the maximum number of iterations to perform during minimization was set to 10,000. Molecular dynamic process was proceeded to obtain conformations with the minimum global energies. The method used in this process was Simulated Annealing. In this method, the molecules were heated at 700 K in a period of 1,000 femtoseconds, they were then cooled down to 200 K in another period of 1,000 femtoseconds to give the stable states from which their final conformations were obtained. The process was run in 5 cycles to figure out different necessary structures. Finally, the energy minimization process was performed one more time and the steric energies of final conformations were specified [72].
Docking was firstly performed on 3 conformations of co-crystallized ligand, including: one separated from the complex (native form, not prepared), one separated from the complex and re-prepared using mentioned appropriate procedure, and one built and prepared from the beginning. The RMSD value between re-docked conformation and the original bound ligand in the co-crystal complex which was ≥ 1.5 Å would indicated the reliability of the binding ability prediction of new ligands. Docking process was performed using FlexX program in BioSolveIT LeadIt. This program applied the flexible-based docking methodology, in which the ligand was treated as a flexible component and the protein was kept rigid during docking process. FlexX used an incremental construction algorithm for the seach of ligand conformations. The base fragment was first placed into the active site by matching interaction geometries between the ligand and protein. Then the remains were gradually built-up in conformity with a set of predefined rotatable torsion angles to account for ligand flexibility. The FlexX utilised empirical scoring functions to score and rank the docking poses [65, 73]. In this study, the docking process was done with following options: The method in which base fragment placed in binding pocket was Triangle Matching; the maximum number of solutions per iteration was set to 1,000; the maximum number of solutions per fragmentation was set to 200; the number of poses to keep for further analysis of interaction was set to 10. The best conformation was the one with the most minus docking score. This score was the total energy emitted from the formation of binding between the molecules and the active site.
The interactions between chalcone molecules and their target were rendered and analyzed in MOE 2008.10 program [74] (hydrogen bonds, π-π interactions, cations-π interactions, ionic interactions). Van der Waals surface interactions were detected by the contact of hydrophilic and lipophilic surfaces of the ligands with those of binding points.
3. RESULTS AND DISCUSSION
Yield: 68%. M.p: 155±1°C. IR (v cm-1, KBr): 1641 (vC=O); 1585 (vC=C aromatic ring); 754 (vC=C1). 1H-NMR (500 MHz, CDCl3, δ ppm): 12.74 (s, 1H, -OH);8.35-8.32 (d, 1H, J = 15.5 Hz, Hβ); 7.95-7.93 (d, 1H, J1 = 8 Hz, J2 = 1.5 Hz, H6,); 7.81-7.79 (dd, 1H, J = 8 Hz, H3,); 7.69-7.66 (d, 1H, J = 15.5 Hz, Hα); 7.56-7.53 (t, 1H, H5,); 7.50-7.49 (d, 1H, J1 = 7.5 Hz, J2 = 1 Hz, H6); 7.41-7.35 (m, 2H, H5 and H4,); 7.8-7.06 (dd, 1H, J1 = 8.5 Hz, J2 = 1 Hz, H3); 6.99-6.96 (t, 1H, H4).
Yield: 74%. M.p: 152±1°C. IR (v cm-1, KBr): 1639 (vC=O); 1564 (vC=C aromatic ring); 683 (vC=C1). 1H-NMR (500 MHz, CDCl3, δ ppm): 12.76 (s, 1H, -OH);7.95-7.93 (d, 1H, J = 8 Hz, H6,); 7.93-7.88 (d, 1H, J = 15.5 Hz, Hβ); 7.67-7.64 (d, 1H, J = 15.5 Hz, Hα); 7.64-7.62 (d, 2H, J = 9 Hz, H2 and H6); 7.56-7.52 (t, 1H, H4,); 7.45-7.44 (d, 2H, J = 8.5 Hz, H3 and H5); 7.08-7.06 (d, 1H, J = 8.5 Hz, H3,); 7.00-6.97 (t, 1H, H5,).
Yield: 74%. M.p: 178±1°C. IR (v cm-1, KBr): 1641 (vC=O); 1583 (vC=C aromatic ring); 710 (vC=C1). 1H-NMR (500 MHz, CDCl3, δ ppm): 12.68 (s, 1H, -OH);8.26-8.23 (d, 1H, J = 15.5 Hz, Hβ); 7.92-7.90 (dd, 1H, J1 = 8 Hz, J2 = 1.5 Hz, H6,); 7.74-7.72 (d, 1H, J = 7.5 Hz, H6); 7.66-7.63 (d, 1H, J = 15.5 Hz, Hα); 7.56-7.53 (t, 1H, J1 = 7 Hz, J2 = 2 Hz, H5,); 7.52 (s, 1H, J = 2 Hz, H3); 7.36-7.34 (dd, 1H, J1 = 9 Hz, J2 = 2 Hz, H5); 7.08-7.06 (d, 1H, J = 7.5 Hz, H3,); 6.99-6.98 (t, 1H, H4,).
Yield: 67%. M.p: 185±1 °C. IR (v cm-1, KBr): 1684 (vC=O); 1576 (vC=C aromatic ring); 746 (vC=C1). 1H-NMR (500 MHz, CDCl3, δ ppm): 12.66 (s, 1H, -O>H); 8.33-8.30 (d, 1H, J = 16 Hz, Hβ); 7.92-7.91 (d, 1H, J1 = 8.5 Hz, J2 = 1 Hz, H6,); 7.70-7.68 (d, 1H, J1 = 8 Hz, J2 = 1.5 Hz, H4); 7.65-7.62 (d, 1H, J = 15.5 Hz, Hα); 7.57-7.55 (dd, 1H, J1 = 8 Hz, J2 = 1.5 Hz, H6); 7.57-7.53 (t, 1H, H5,); 7.33-7.29 (t, 1H, H4,); 7.8-7.06 (dd, 1H, J1 = 8 Hz, J2 = 1 Hz, H3,); 7.00-6.96 (t, 1H, H5).
Yield: 71%. M.p: 113±1°C. IR (v cm-1, KBr): 1638 (vC=O), 1558 (vC=C aromatic ring), 1171 (vC-O). 1H-NMR (500 MHz, MeOD, δ ppm): 8.20-8.18 (d, 1H, J = 15.5 Hz, Hβ); 8.06 8.4 (d, 1H, J = 8 Hz, H6,); 7.81-7.78 (d, 1H, J = 15.5, Hα); 7.75-7.73 (d, 1H, J = 9.5 Hz, H3,); 7.51-7.47 (m, 1H, H5°); 6.98-6.95 (m, 1H, H4,); 6.95-6.93 (d, 1H, J = 8 Hz, H6); 6.62-6.60 (ds, 2H, J = 7.5 Hz, H3 and H5); 3.94 (s, 3H, Ar--OCH3); 3.86 (s, 3H, Ar-OCH3).
Yield: 48%. M.p: 107±1°C. IR (v cm-1, KBr): 3279 (vO-H), 1639 (vC=O), 1607 (vC=C aromatic ring), 1117 (vC-O). 1H-NMR (500 MHz, CDCl3, δ ppm): 12.83 (s, 1H, -OH phenol); 8.228.18 (d, 1H, J = 15.5 Hz, Hβ); 7.92-7.91 (d, 1H, J = 1.5 Hz, 8 Hz, H6,); 7.77-7.74 (d, J = 15.5, 1H, Hα); 7.51-7.48 (t, 1H, H4,); 7.29=-7.27 (d, 1H, J1 = 1 Hz, J2 = 8 Hz, H6); 7.12-7.09 (t, 1H., H5); 7.04-7.02 (d, 1H, J1 = 1 Hz, J2 = 8 Hz, H6); 7.00-6.99 (d, 1H, J1 = 1 Hz, J2 = 8 Hz, H3,); 3.92 (s, 3H, -OCH3); 3.90 (s, 3H, -OCH3).
Yield: 67%. M.p: 157±1 °C. IR (v cm-1, KBr): 1636 (vC=O); 1568 (vC=C aromatic ring), 1155 (vC-O). 1H-NMR (500 MHz, MeOD, δ ppm): 8.25-8.24 (d, 1H, J = 8 Hz, H6,); 7.90 (s, 2H, 15 Hz, Hα and Hβ), 7.57-7.54 (t, 1H, J = 7.5 Hz, H5,), 7.16 (s, 2H, H2 and H6), 7.04-7.00 (t, 1H, J = 7.5 Hz, H4,); 7.02-7.00 (d, 1H, J = 9.5 Hz, H3,); 3.95 (s, 6H, Ar-OCH3); 3.85 (s, 3H, Ar-OCH3).
Yield: 87%. M.p: 180±1 °C. IR (v cm-1, KBr): 1622 (vC=O), 1599 (vC=C aromatic ring), 1155 (vC-O). 1H-NMR (500 MHz, CDCl3, δ ppm): 13.17 (s, 1H, -OH phenol); 7.937.90 (d, 1H, J = 15.5 Hz, Hβ); 7.93-7.91 (d, 1H, J1 = 1.5 Hz, J2 = 7.5 Hz, H6); 7.58-7.56 (d, 2H, J = 8 Hz, H2, and H6,); 7.47-7.44 (d, 1H, J = 15 Hz, Hα); 7.47-7.44 (t, 1H, H4); 7.02 7.0 (d, 1H, J = 8 Hz, H3); 6.9°4-6.90 (t, 1H, H4,); 6.71-6.69 (d, 2H, J1 = 2 Hz, J2 = 8 Hz, H3 and H5); 3.06 (s, 6H, -CH3).
Yield: 58%. M.p: 133±1 °C. IR (v cm-1, KBr): 1638 (vC=O), 1582 (vC=C aromatic ring), 1269 (vC-O). 1H-NMR (500 MHz, CDCl3, δ ppm): 13.44 (s, 1H, -OH phenol); 8.17-8.14 (d, 1H, J = 15,5 Hz, Hβ); 7.83-7.81 (d, 1H, J1 = 2 Hz, J2 = 9 Hz, H6,); 7.68-7.65 (d, 1H, J = 16 Hz, Hα); 7.27-7.26 (d, 1H, J1, = 1.5 Hz, J2, = 8 Hz, H6); 7.11-7.08 (t, 1H, J = 8 Hz, H5); 7.00-6.97 (d, 1H, J1 = 1,5 Hz, J2 = 8 Hz, H5,); 6.49-6.48 (d, 1H, J1 = 2.5 Hz, J2 = 8 Hz, H3); 6.47 (s, 1H, H3,); 3.91 (s, 3H, -OCH3), 3.90 (s, 3H, -OCH3); 3.86 (s, 3H, -OCH3).
Yield: 62%. M.p: 170±1 °C. IR (v cm-1, KBr): 1634 (vC=O), 1565 (vC=C aromatic ring), 1126 (vC-O). 1H-NMR (500 MHz, CDCl3, δ ppm): 13.50 (s, 1H, -OH phenol); 7.86-7.82 (d, 1H, J = 15.5 Hz, Hβ); 7.84-7.82 (d, 1H, J = 8 Hz, H6,); 7.44-7.41 (d, 1H, J = 15.5 Hz, Hα); 7.26-7.24 (d, 1H, J1, = 2 Hz, J2, = 8.5 Hz, H6); 7.16 (d, 1H, J = 2 Hz, H3,); 6.91-6.90 (d, 1H, J = 8 Hz, H5); 6.50-6.47 (d, 1H, J1 = 2.5 Hz, J2 = 8.5 Hz, H5,); 6.47 (s, 1H, J = 2 Hz, H2); 3.96 (s, 3H, -OCH3); 3.93 (s, 3H, -OCH3), 3.86 (s, 3H, -OCH3).
Yield: 63%. M.p: 123±1 °C. IR (v cm-1, KBr): 1643 (vC=O), 1574 (vC=C aromatic ring), 1202 (vC-O). 1H-NMR (500 MHz, CDCl3, δ ppm): 13.43 (s, 1H, -OH phenol); 7.84-7.82 (d, 1H, J1 = 2 Hz, J2 = 9 Hz, H6,); 7.82-7.78 (d, 1H, J = 16 Hz, Hβ); 7.46-7.43 (d, 1H, J = 16 Hz, Hα); 6.87 (s, 2H, H2 and H6); 6.50-6.48 (d, 1H, J1 = 2.5Hz, J2 = 9 Hz, H5); 6.48 (s, 1H, H3); 3.93 (s, 6H, -OCH3 x 2); 3.91 (s, 3H, -OCH3), 3.86 (s, 3H, -OCH3)
Yield: 68%. M.p: 143±1 °C. IR (v cm-1, KBr): 1635 (vC=O), 1566 (vC=C aromatic ring), 1204 (vC-O). 1H-NMR (500 MHz, CDCl3, δ ppm): 13.33 (s, 1H, -OH phenol); 7.84-7.80 (d, 1H, J = 15.5 Hz, Hβ); 7.81-7.79 (d, 1H, J = 8.5 Hz, H6); 7.58-7.56 (d, 2H, J1 = 2 Hz, J2 = 8.5 Hz, H2, and H6,); 7.55-7.52 (d, 1H, J = 15.5 Hz, Hα); 7.41-7.39 (d, 2H, J1 = 2.5 Hz, J2 = 8.5 Hz, H3, and H5,); 6.50-6.47 (d, 1H, J1 = 2,5 Hz, J2 = 8,5 Hz, H5); 6.48 (s, 1H, H3); 3.86 (s, 3H, -OCH3).
Yield: 55%. M.p: 132±1 °C. IR (v cm-1, KBr): 1549 (vC=O), 1504 (vC=C aromatic ring), 1119 (vC-O). 1H-NMR (500MHz, DMSO, δ ppm): 13.79 (s, 1H, -OH phenol); 7.91-7.88 (d, 1H, J = 15.5 Hz, Hβ); 7.80-7.77 (d, 1H, J = 16 Hz, Hα); 7.65-7.63 (d, 1H, J = 8.5 Hz, H6); 6.65 (d, 1H, J = 2 Hz, H3); 6.63-6.61 (dd, 1H, J1 = 2,5 Hz, J2 = 8.5 Hz, H5); 6.14 (d, 1H, J = 2 Hz, H3,); 6.11 (d, 1H, J = 2 Hz, H5,); 3.90 (s, 6H, -OCH3); 3.84 (s, 3H, -OCH3); 3.82 (s, 3H, -OCH3).
Yield: 66%. M.p: 130±1 °C. IR (v cm-1, KBr): 1622 (vC=O), 1549 (vC=C aromatic ring), 1304 (vC-O). 1H-NMR (500MHz, DMSO, δ ppm): 13.37 (s, 1H, -OH phenol); 7.67-7.63 (d, 1H, J = 15.5 Hz, Hβ); 7.62-7.59 (d, 1H, J = 15.5 Hz, Hα); 7.30-7.28 (dd, 2H, J1 = 7 Hz, J2 = 2 Hz, H2 and H6); 7.03-7.02 (d, 1H, J = 8.5 Hz, H5); 6.16-6.15 (d, 1H, J = 2.5 Hz, H3,); 6.12 (d, 1H, J = 2.5 Hz, H5,); 3.89 (s, 3H, -OCH3); 3.83 (s, 3H, -OCH3); 3.82 (s, 6H, -OCH3).
Yield: 72%. M.p: 134±1 °C. IR (v cm-1, KBr): 1638 (vC=O), 1593 (vC=C aromatic ring), 1113 (vC-O). 1H-NMR (500MHz, DMSO, δ ppm): 13.54 (s, 1H, -OH phenol); 7.82-7.78 (d, 1H, J = 16 Hz, Hβ); 7.76-7.73 (d, 1H, J = 15.5 Hz, Hα); 7.50 (d, 1H, J = 8.5 Hz, H6); 6.92-6.91 (d, 1H, J = 9 Hz, H5); 6.16-6.15 (d, 1H, J = 2.5 Hz, H3,); 6.12 (d, 1H, J = 2.5 Hz, H5,); 3.89 (s, 3H, -OCH3); 3.86 (s, 3H, -OCH3); 3.85 (s, 3H, -OCH3); 3.82 (s, 3H, -OCH3); 3.78 (s, 3H, -OCH3).
Yield: 69%. M.p: 138±1 °C. IR (v cm-1, KBr): 1630 (vC=O), 1568 (vC=C aromatic ring), 1290 (vC-O). 1H-NMR (500MHz, DMSO, δ ppm): 13.31 (s, 1H, -OH phenol); 7.77-7.73 (t, 3H, J1, = 8.5 Hz, J2, = 7.5 Hz, J3 = 16 Hz, Hβ, H3, H4); 7.64-7.61 (d, 1H, J = 16 Hz, Hα); 7.52-7.50 (d, 2H, J = 8.5 Hz, H2 and H5); 6.17-6.16 (d, 1H, J = 2 Hz, H3,); 6.14-6.13 (d, 1H, J = 2.5 Hz, H5,); 3.90 (s, 3H, -OCH3); 3.83 (s, 3H, -OCH3).
Yield: 66%. M.p: 135±1 °C. IR (v cm-1, KBr): 3420 (vNH2); 1653 (vC=O) (vC=C somatic ring); 687 (vC-C1). 1H-NMR (500 MHz, CDCl3, δ ppm): 8.14-8.12 (d, 1H, J = 8.5 Hz, H3); 7.96-7.94 (d, 2H, J1, = 9 Hz, J2 = 2 Hz, H2 and H6); 7.77-7.75 (m, 1H, H5); 7.57-7.54 (d, 1H, J = 15.5 Hz, Hβ); 7.53-7.46 (m, 1H, H4); 7.46-7.44 (d, 1H, J = 8 Hz, H6); 7.34-7.31 (d, 1H, J= 15.5 Hz, Hα); 6.73-6.71 (dd, 2H, J1, = 7 Hz, J2 = 2 Hz, H3 and H5,); 4.23 (s, 2H, -NH2).
Yield: 70%. M.p: 114±1 °C. IR (v cm-1, KBr): 3460 (vNH2); 1645 (vC=O); 1605 (vC=C somatic ring); 746 (vC-C1). 1H-NMR (500 MHz,, CDCl3 δ ppm): 7.96-7.94 (dd, 2H, J1 = 7 Hz, J2 = 2 Hz, H2, and H6,); 7.76-7.73 (d, 1H, J = 15.5 Hz, Hβ); 7.59 (dd, 2H, J1 = 6.5 Hz, J2 = 1.5 Hz, H3, and H5,); 7.55-7.52 (dd, 1H, J = 15.5 Hz, Hα); 7.61-7.52 (d, 2H, J1, = 7 Hz, J2 = 2 Hz, H2 and H6); 7.41-7.39 (dd, 2H, J1 = 7 Hz, J2 = 2 Hz, H3 and H5); 4.17 (s, 2H, -NH2).
Yield: 76%. M.p: 174±1 °C. IR (v cm-1, KBr): 3489 (vNH2); 1634 (vC = O); 1506 (vC=C aromatic ring). 1H-NMR (500 MHz, DMSO, δ ppm): 8.30-8.28 (d, 2H, J = 9 Hz, H3 and H5); 8.13-8.12 (d, 2H, J = 8.5 Hz, H2 and H6); 8.08-8.05 (d, 1H, J = 15.5 Hz, Hβ); 7.96-7.95 (d, 2H, J = 8.5 Hz, H2, and H6,); 7.70-7.67 (d, 1H, J = 15.5 Hz, Hα); 6.64-6.62 (d, 2H, J = 9 Hz, H3, and H5,); 6.24 (s, 2H, -NH2).
Yield: 60%. M.p: 185±1 °C. IR (v cm-1, KBr): 1666 (vC=O); 1541 (vC=C aromatic ring). 1H-NMR (500 MHz, DMSO, δ ppm): 8.88-8.87 (m, 1H, H4,); 8.64-8.62 (d, 1H, H6,); 8.54-8.52 (m, 1H, H2,); 8.32-8.30 (d, 2H, J = 7.5 Hz, H3 and H5); 8.24 (d, 2H, J = 8, H2 and H6); 8.22-8.21 (d, 1H, J = 15.5, Hβ), 7.94-7.92 (d,1H, J = 16 Hz, Hα), 7.91-7.89 (d, 1H, H5,).
Inhibitory activities against AChE of the chalcone derivatives S1-S20 were determined by the Ellman’s method, with galantamine as a reference compound. The IC50 values for AChE inhibition are indicated in Table 2. Results of AChE inhibitory assay showed that in comparision with the control substance galantamine, synthesized chalcone compounds had weaker activities. And based on their activities, they could be divided into 3 groups: those with strong activities including S1, S2, S3, S4, S5, S17 whose IC50 values were from 36.10 pM (S17) to 92.42 μM (S1), those with average activities including S9, S10, S16 whose IC50 values were from 129.90 (S10) to 213.14 (S16), and the rest with weak activities. The results also indicated that the derivative S17 was the most potent compound.
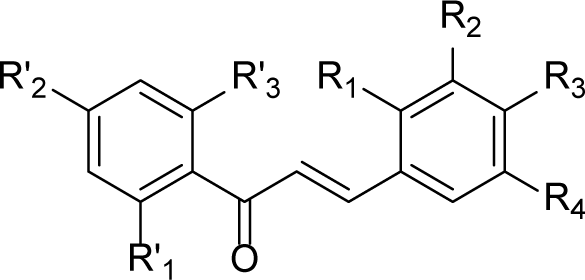
The Lineweaver-Burk plots (Fig. 2) describing the AChE inhibition by S17 showed both increasing slopes and increasing intercepts with higher inhibitor concentrations. This pattern indicated a mixed-type inhibition of enzyme and it was in agreement with the results of studies carried out by Liu et al [51, 52, 55] on the chalcone analogs of S17. These results also revealed that compound S17 and the analogs bound to both catalytic active site and the peripheral anionic site of AChE, which was elucidated by molecular modeling studies.
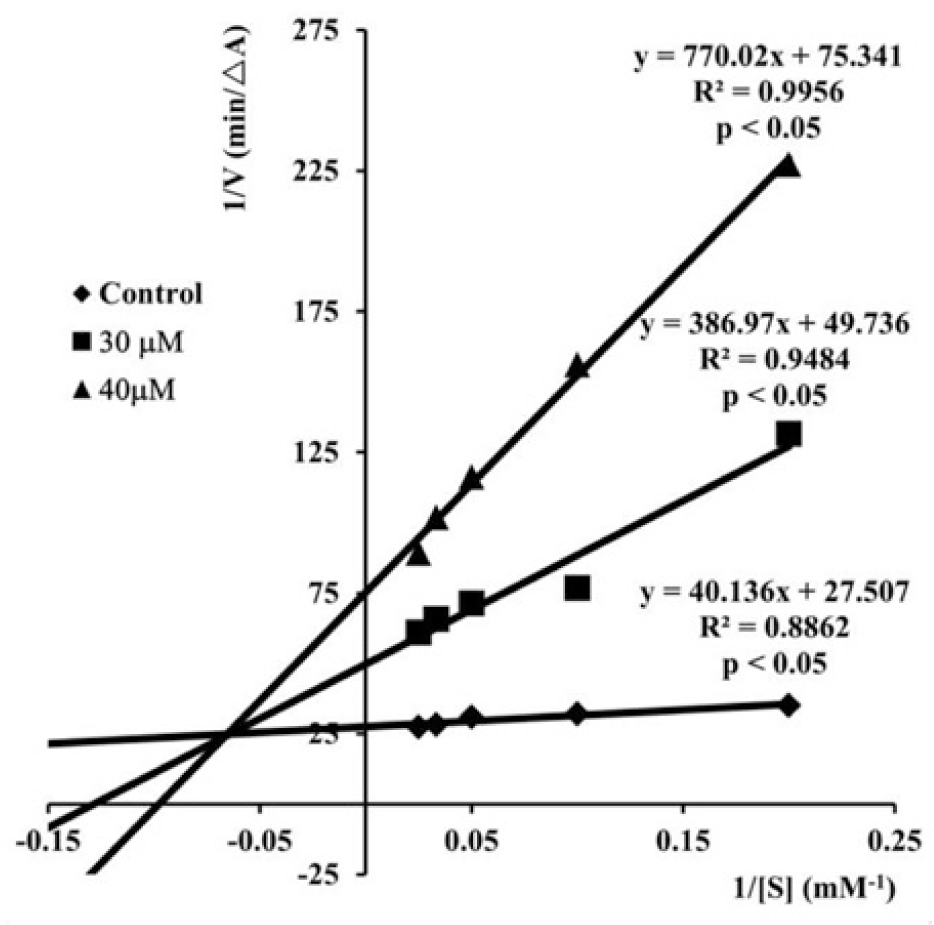
The rather tight binding of galantamine to TcAChE came from many interactions with the protein. The model of interaction between galantamine and its target (Fig. 3) indicated that this compound could make 3 interaction with the catalytic site, including 2 hydrogen bonds with Ser220 and His440 of the catalytic triad, 1 strong hydrogen bond between the hydroxyl group with Glu199 of the ‘anionic’ subsite with the distance of about 1.5 Â. In addion there was one arene-cation interaction between galantamine and residue Phe330 of the ‘anionic’subsite in the enzyme’s active site. These were important bonds which contributed to the potent activity of galantamine and could be considered in the interactions of studied chalcones.
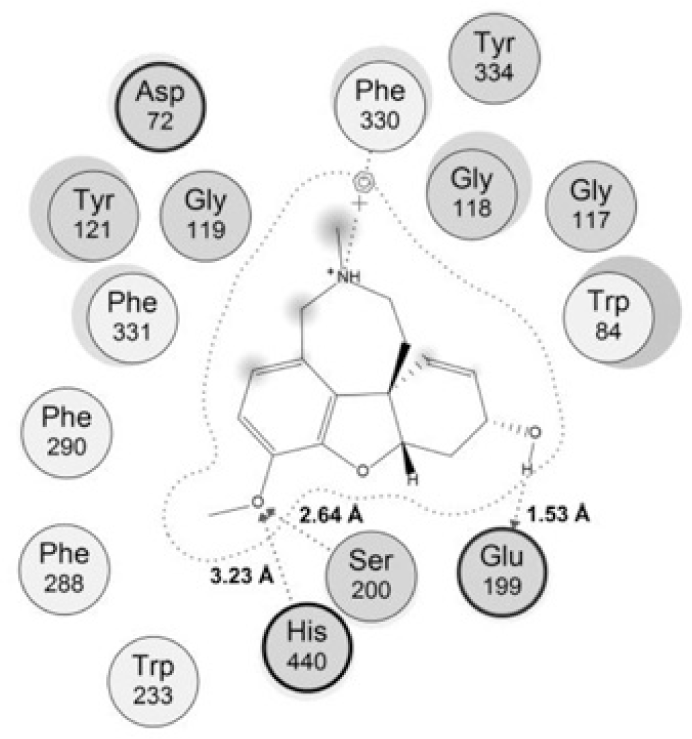
Re-docking results of galantamine showed that interactions made by re-docked conformations with the active site were resemble those of the original bound ligand in 1DX6 (Fig. 4). The RMSD values of re-docked conformations were < 1.5 Å (Table 3) indicated that the molecular model could be applied to explain the interactions of new ligands with the active site.
| Ligand | RMSD (Å) |
|---|---|
| 1 | 0.4912 |
| 2 | 0.5184 |
| 3 | 0.5021 |
The docking process was performed successfully with all 20 chalcone compounds. Docking scores are listed in Table 4. There was a good correlation between docking scores and bioactivities of studied chalcone compounds. This correlation is indicated in Fig. 5.
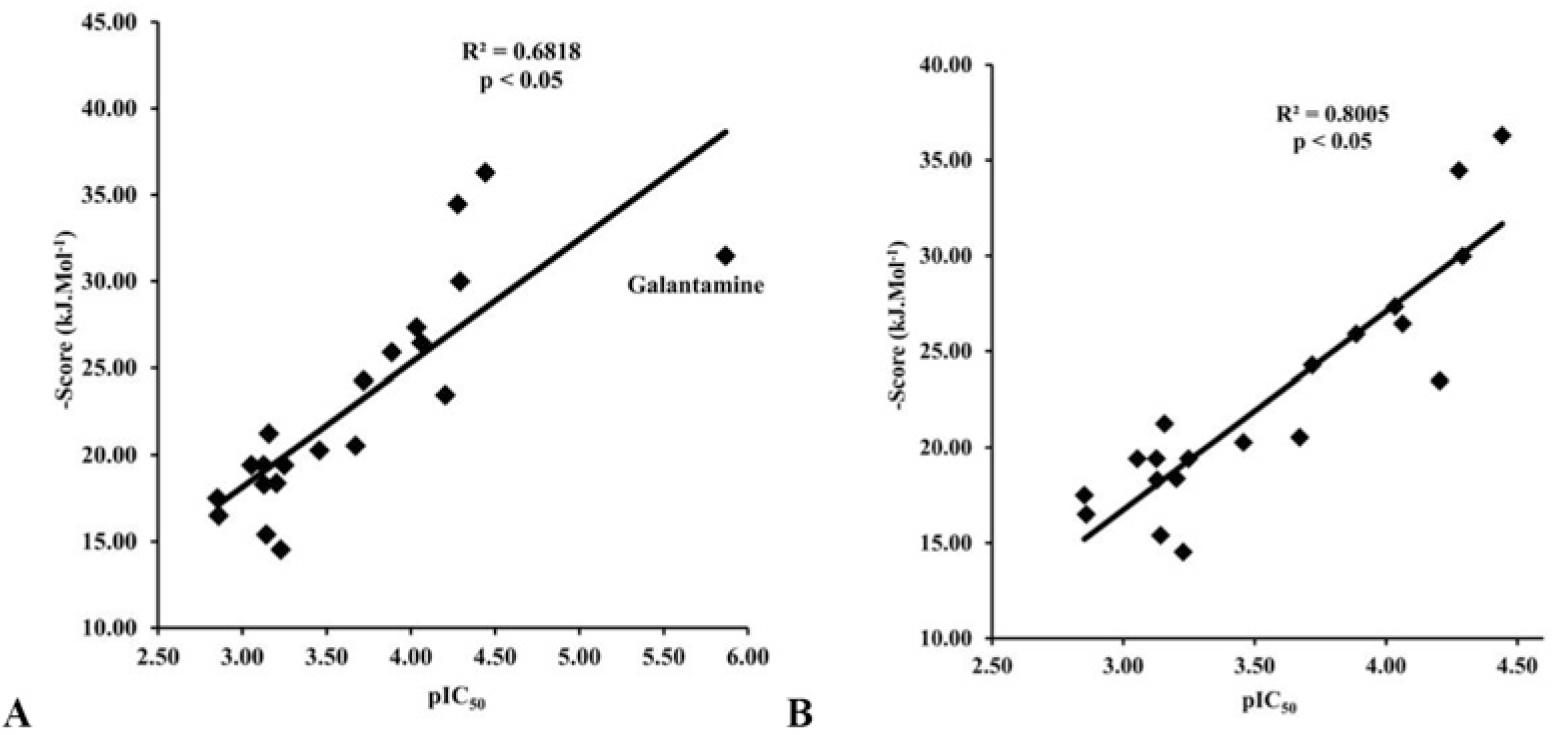
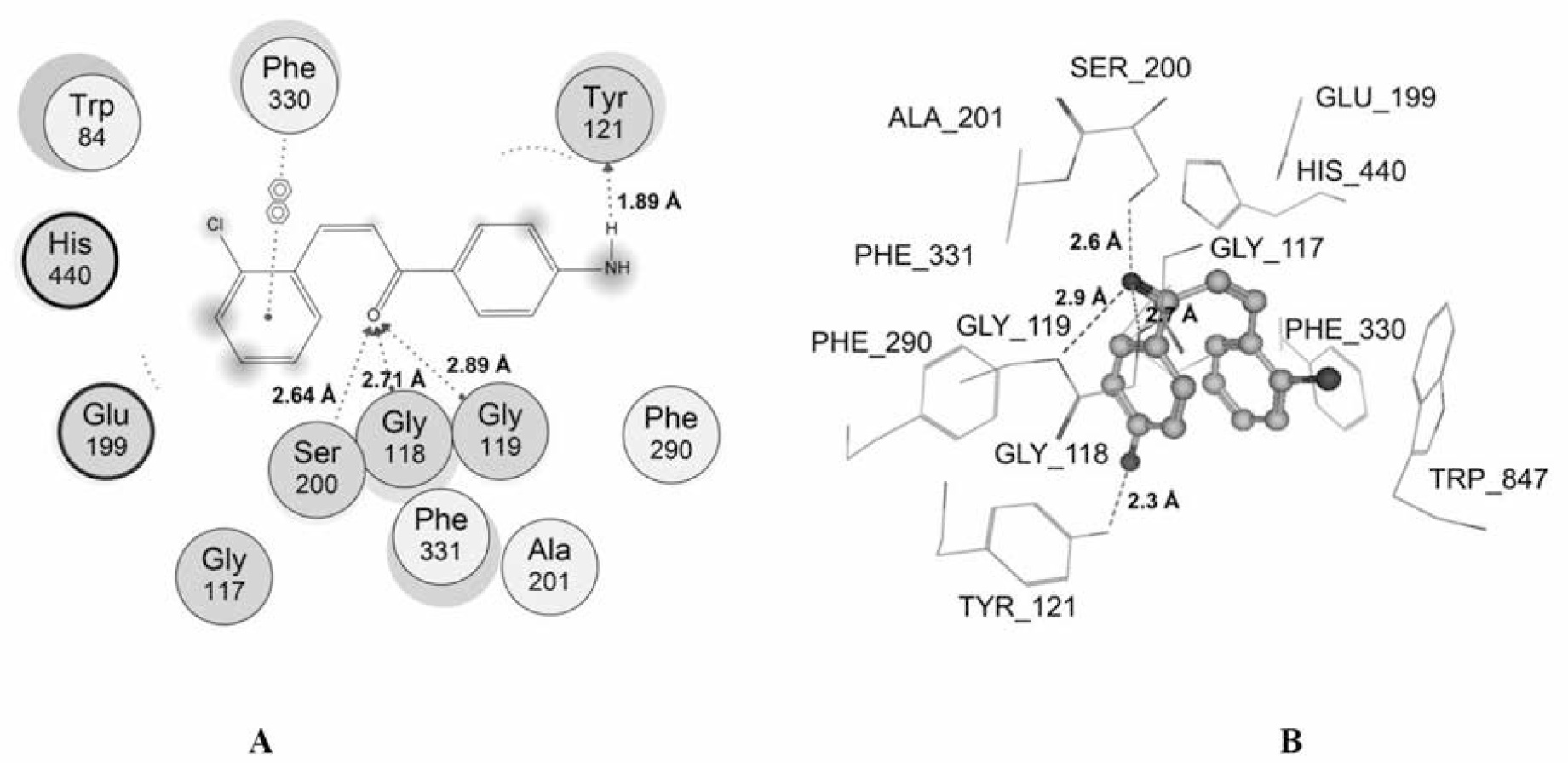
Among the studied compounds, S17 showed the strongest interaction with its target. This compound bound to both catalytic active site and the peripheral anionic site of AChE. This was similar to chalcone analogs in the studies of Liu et al [51, 52, 55], but S17 interacted with the different residues. S17 made 4 interactions with the catalytic site, including 1 hydrogen bond with Ser200 of the catalytic triad, 2 hydrogen bonds with the “oxyanion” pocket (Gly118 and Gly119), and 1 arene-arene interaction with Phe330 of anionic subsite. Moreover, S17 could make 1 strong hydrogen bond with the important residue of the peripheral site Trp121 (Fig. 6). The docking score of S17 is -36.29 kJ.mol-1, which was the most minus value of the chalcone compounds in the docking process. All of these could give the explanation for the highest AChE inhibitory activity of S17 in the studied structures. However, this compound was less potent than galantamine, and the reason for this could arise from the fact that galantamine interacted with catalytic site of AChE more effectively with 2 hydrogen bonds with catalytic triad and 1 strong hydrogen bond with “oxyanion” pocket. In addition, a low entropy cost for binding due to the rigid nature of the galantamine structure which did not occur with S17 molecule also gave a further explanation.
4. CONCLUSIONS
By applying Claisen-Schmidt condensation method, 20 chalcone derivatives were synthesized. These compounds were purified and determined their physiochemical parameters including IR and 1H-NMR spectra. Among those which were determined for in vitro acetylcholinesterase inhibitory activities, six had IC50 values below 100 μM; three had the IC50 values in the range of 100 - 250 μM and all the rest had IC50 values above 250 μM or not detected. Compound S17 (4’-amino-2-chlorochalcone) showed the highest AChE inhibitory activity with IC50 value of 36.10 μM. It could be considered to use as a lead compound for the studies on AChE inhibitory activity of chalcone compounds. Together with the results of acetylcholinesterase inhibitory assay, the molecular docking studies gave some elucidation for the binding modes of chalcones to the active site of AChE. The information could be valuable for further investigation and would be useful in later research on the design and synthesis new acetylcholinesterase inhibitors.