






1. INTRODUCTION
Central neurocytoma (CN) is a rare tumor that was first individualized by Hassoun et al. in the 1980s [1]. According to the Central brain Tumor Registry of the United States, representing approximately 100% of the US population, the annual incidence rate of CN was 0.022 per 100 000 between 2006 and 2014 [2]. Central neurocytoma until 2020 has 500 known cases worldwide [3], mostly comprising of case reports and short series from individual specialties. This kind of tumor typically occurs in the lateral ventricular near the foramen of Monro. It is composed of uniform round cells that show evidence of neuronal differentiation on immunohistochemistry and electron microscopy. CN has been clinically misdiagnosed as ependymomas of the foramen of Monro or intraventricular oligodendrogliomas due to overlapping radiological and microscopic features [4]. Nonetheless, the usual benign clinical course and the neuronal differentiation of central neurocytoma designates it as a distinct entity [4]. Because of its rarity, the diagnosis of this neoplasm remains controversial and may be tricky in some instances. Herein, we report a case of central neurocytoma in a young middle-age male patient that was initially diagnosed as ependymoma. We also review clinical, radiological, histopathological, and immunohistochemical characteristics for CN comprehensive diagnosis.
2. CASE PRESENTATION
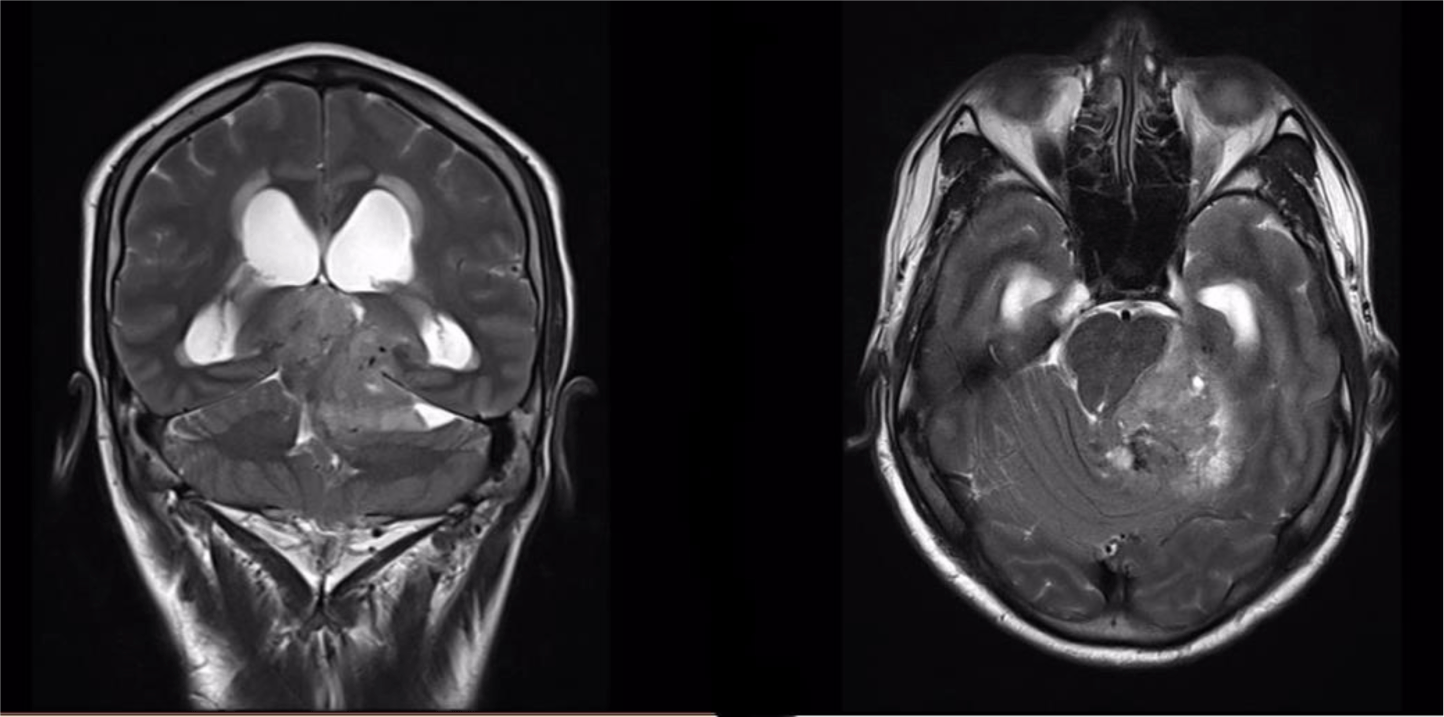
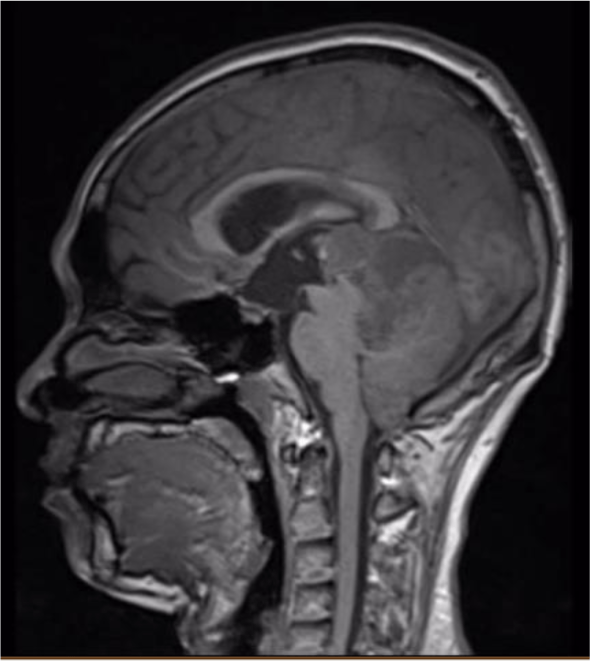
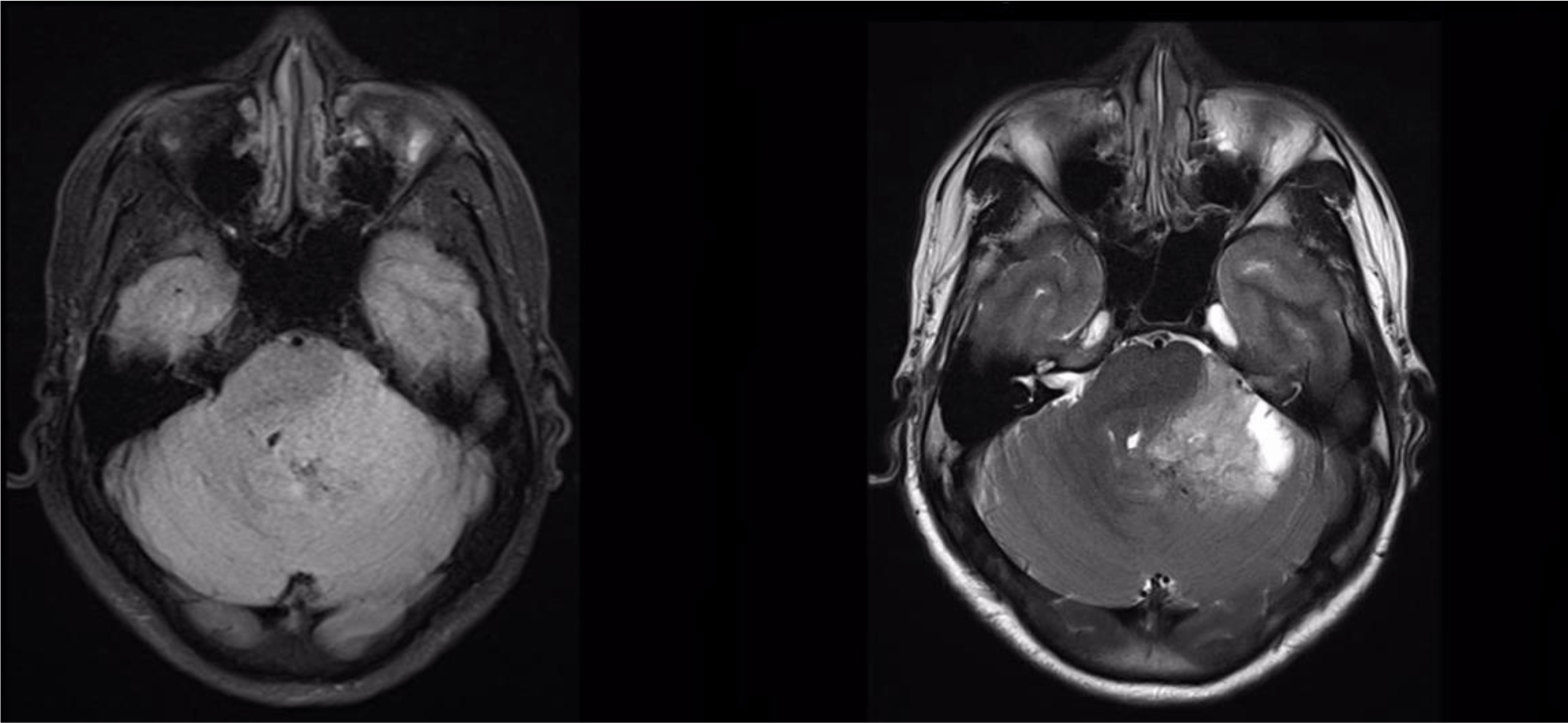
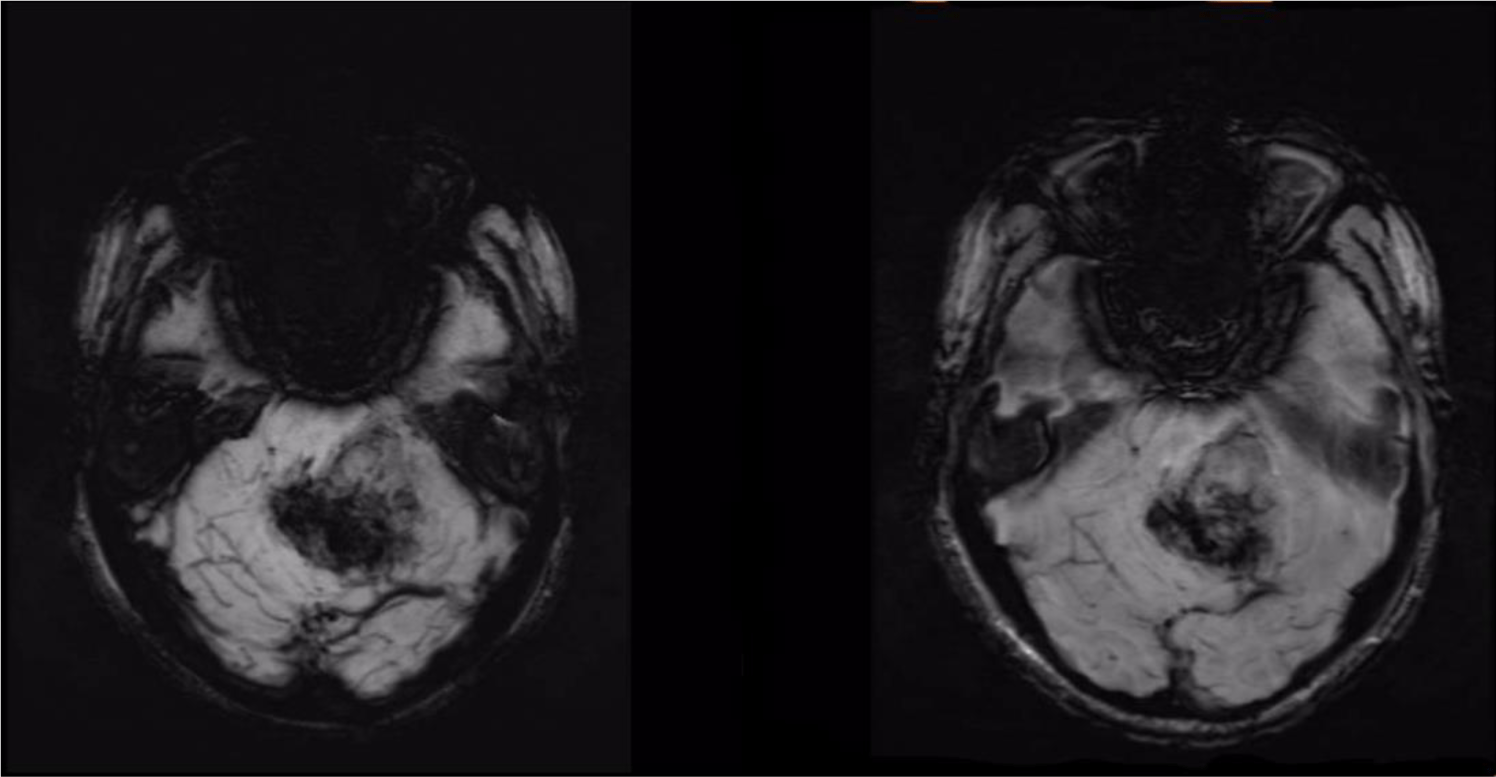
A 32-year-old man suffered from constant headaches and frequent vertigo for over a month, then presented with sudden, severe vomiting and vertigo. Physical examination showed neither papilledema nor neurological deficits. Magnetic resonance imaging (MRI) revealed T1 hypointense, T2 hyperintense fairly-marginated inhomogeneously enhancing mass of size 42x50x55 mm in the left cerebellopontine angle, which partially located in the third ventricle and extended through the foramina of Luschka and the aqueduct of Sylvius into the fourth ventricle (Figure 1-3), which showed heterogeneous gadolinium enhancement with scattered necrosis, hemorrhage, and calcifications (Figure 4 & Figure 5). The lesion compressed bilateral hippocampi and the left cerebellar hemisphere, pushed the brainstem and the cerebellar vermis to the right, obstructed the cerebral aqueduct, and caused supratentorial ventricular system dilatation. The imaging and clinical symptoms suspected an ependymoma with ventricular dilatation. The patient underwent external ventriculostomy and microsurgical tumor removal.
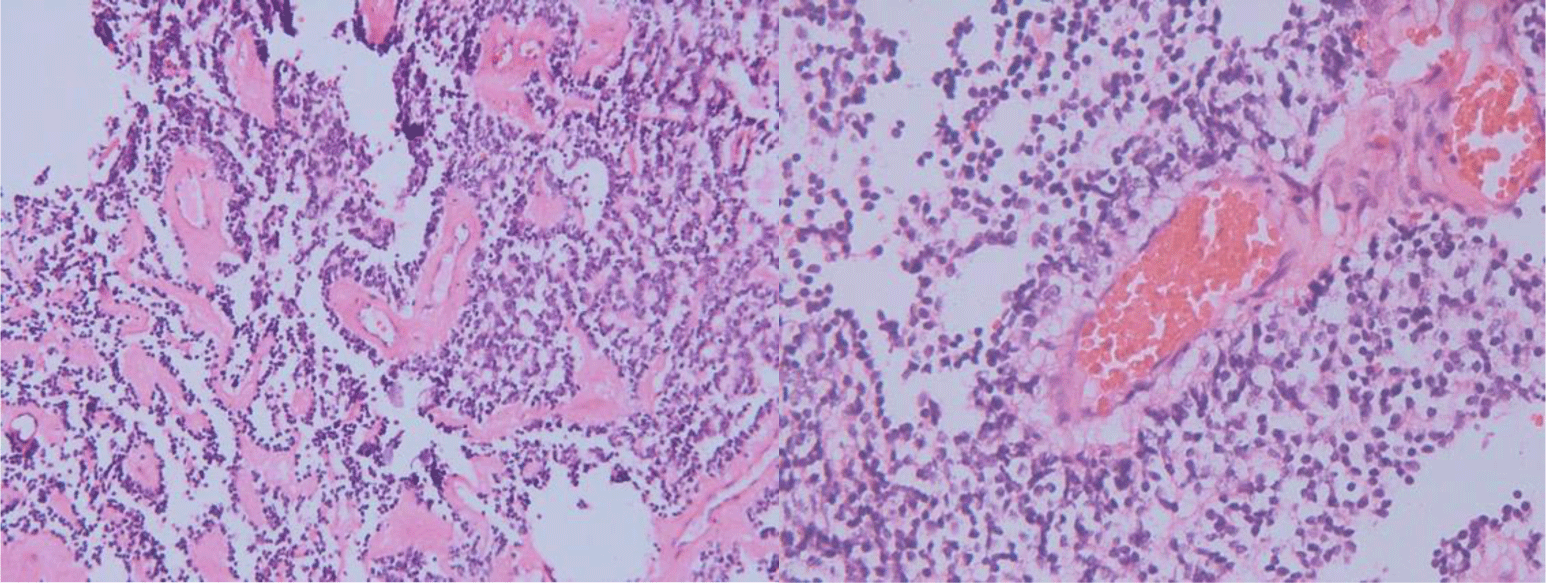
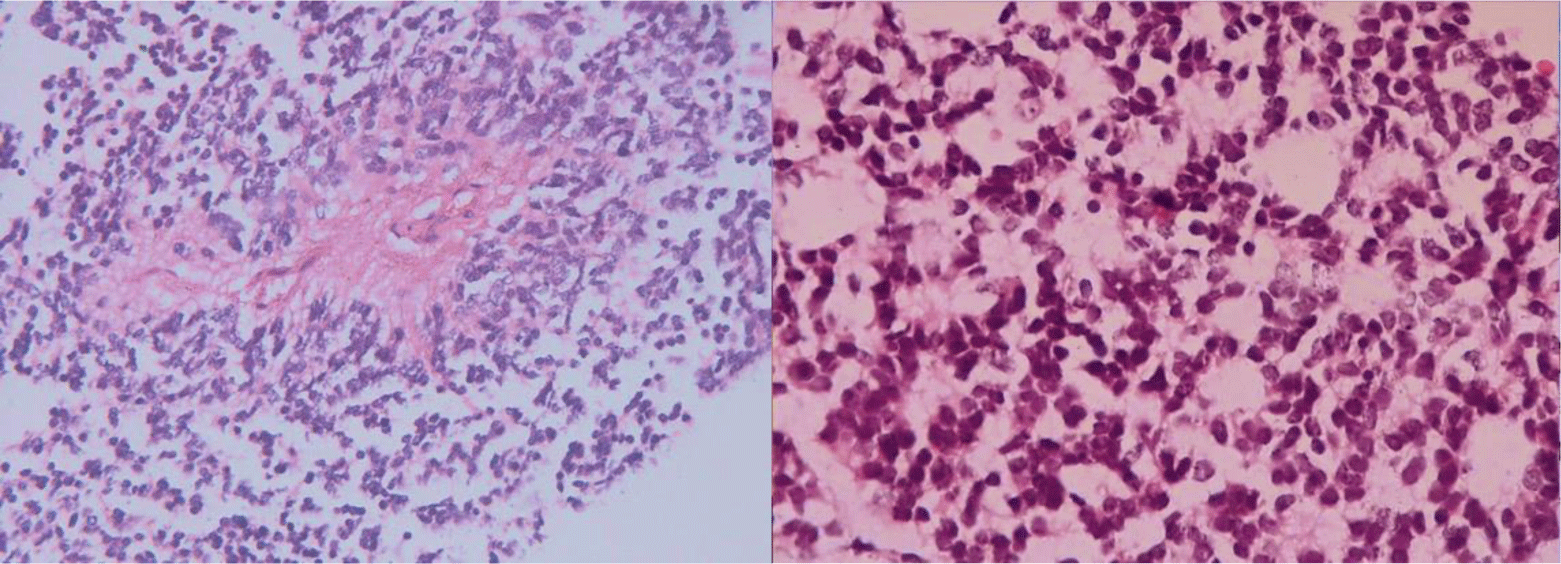
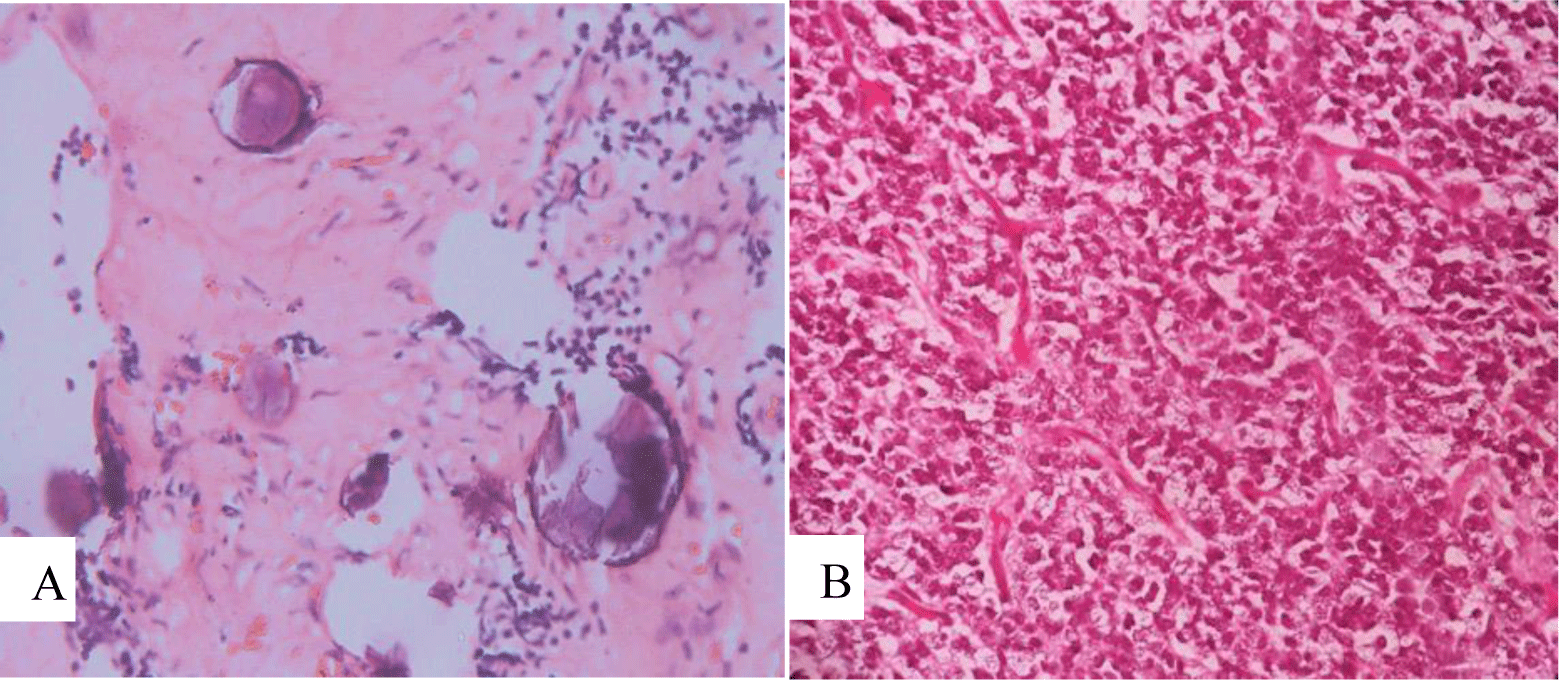
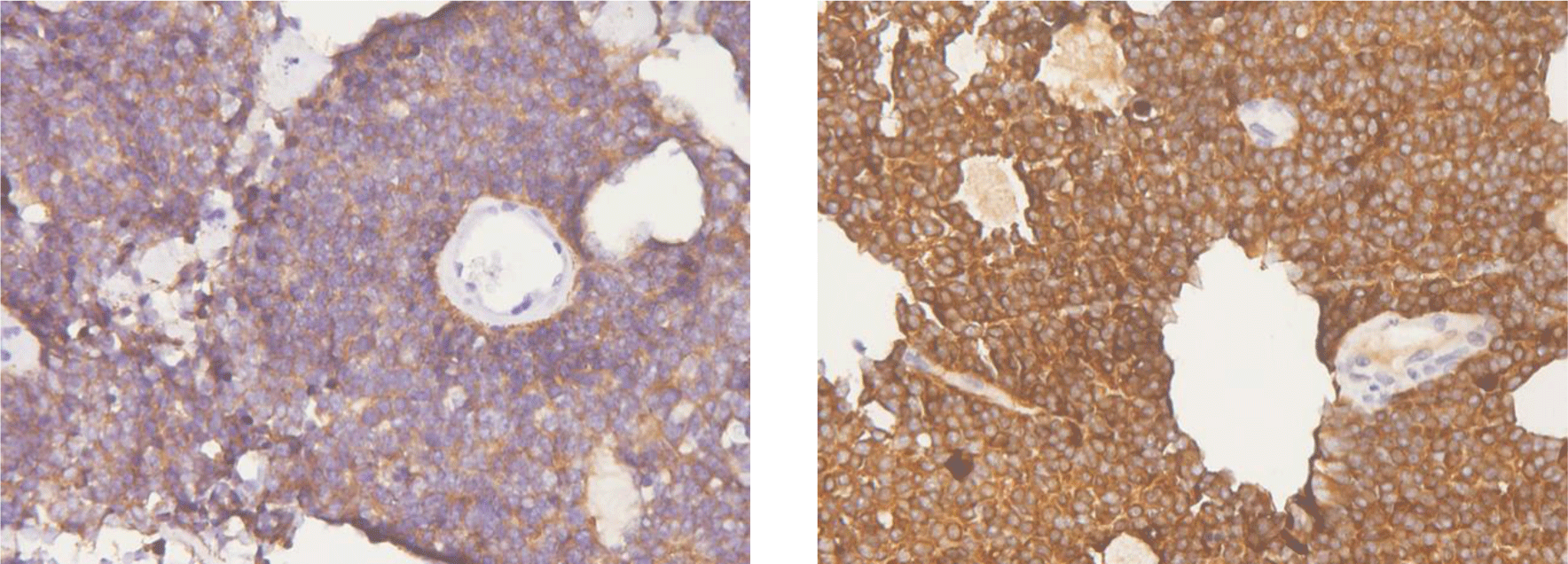
Microscopic features showed uniform, small-to-medium-sized cells with rounded to oval nuclei, finely stippled chromatin, and inconspicuous nucleoli, along with scant cytoplasm, which arranged radially around small central blood vessels as a pseudo-rosette pattern mimicking ependymoma (Figure 6). Fibrillary nucleus-free areas irregular rosette in pineocyomas are also focally presented (Figure 7). Microcalcifications were scattered (Figure 8A). Arborizing capillary blood vessels were detectable, but microvascular proliferation was absent (Figure 8B). Necrosis, mitoses, and abnormal mitotic figures were not found. Immunohistochemical staining showed diffuse and strong cytoplasmic expression for synaptophysin and NSE, whereas indisputably negative for GFAP, CK, EMA, and vimentin.
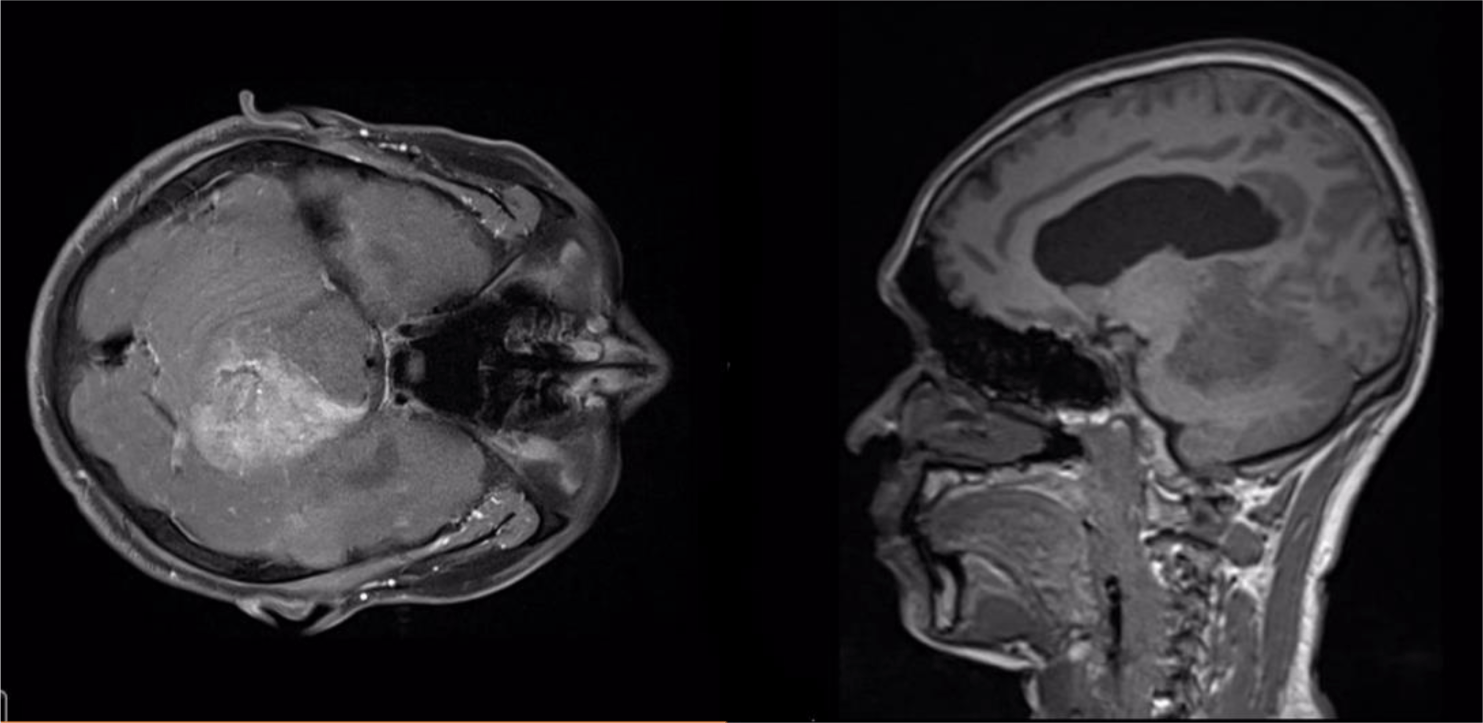
After surgery, intraventricular fluid accumulation and ventricular dilatation did not regress. A brain computed topography (CT) scan two weeks later showed profuse fluid accumulation and dilation of the supratentorial ventricular system, slight fluid accumulation around the left cerebellar hemisphere, brainstem, and also in the left cerebellopontine angle; the left cerebellar hemisphere and brainstem were still compressed. The patient suffered from left-sided hemiplegia, right abducens nerve palsy, and lack of consciousness recovery.
3. DISCUSSION
In literature, CN usually affects young adults and is located in the lateral and third ventricles, obstructing the liquor drainage-headaches and other symptoms of raised intracranial pressure, rather than distinct neurological deficits [4]. Our patient had a similar sign of a typical neurocytoma with sudden onset. Clinical features such as severe headaches, vertigo, and projectile vomiting can appear or surge dramatically due to intraventricular hemorrhage within the tumor, which causes severe obstructive hydrocephalus. In this case, MRI showed a relatively large mass lesion, which was partially located in the third ventricle and extended through the foramina of Luschka and the aqueduct of Sylvius into the fourth ventricle. The tumor had TW1-intermediate intensity, FLAIR-hyperintensity, and heterogeneous gadolinium enhancement with necrosis areas and microcalcifications, which corresponded with the radiological features of CN [4]. Images of the blood accumulation and the ventricular dilatation confirmed tumor hemorrhage and obstructive hydrocephalus complication. Imaging differential diagnosis of CN depends on involved sites and patient’s age [5]. Tumors involve lateral ventricle in young adults include oligodendrogliomas, subependymal giant cell astrocytomas, ependymomas, and low grade or pilocytic astrocytomas [5]. Astrocytomas and ependymomas may involve other sites of the ventricular system, but usually without intratumoral calcifications [5].
Macroscopically, neurocytoma presents as a soft, ovoid, lobulated, or nodular mass that is generally well circumscribed. The neoplasm is usually grayish, resembling the grey matter, with areas of hemorrhage [6]. In our case, the tumor was 42x50x55 mm in size, grayish, and hemorrhagic.
Microscopically, typical central neurocytoma is well-differentiated with the benign histological feature, which is composed of uniform cells with rounded nuclei, finely spackled chromatin, and present variable nucleoli, along with scant eosinophilic cytoplasm and in some areas has small cells with perinuclear halos, which resemble oligodendroglioma [7]. Tumor cells are usually closely arranged but may also be set within a background of finely filamentous stroma with neuropil-like quality. In some places, tumors display high cellular density alternating with fibrillary/acellular networks [6]. The latter growth pattern mainly appears perivascular and has a fine fibrillary neuropil matrix mimicking pseudo-rosettes of ependymoma. In many cases, thin-walled arborizing blood vessels and foci of calcification are readily identified [4]. Anaplastic histological features are characterized by brisk mitotic activity, microvascular proliferation, and necrosis. And tumors with these features are so-called atypical central neurocytoma. The terminology is also applied for CN with a Ki-67 proliferation index above 2 or 3%, even if no anaplastic features are found [4]. In our case, microscopic features revealed cytological characteristics of a typical CN. Tumor cells are arranged radially around small central blood vessels forming a perivascular pseudo-rosette growth pattern, mimicking ependymomas. Although microvascular proliferation was presented, other anaplastic histological features such as necrosis and brisk mitotic activity were absent.
The neuronal differentiation of tumor cells was confirmed by a typical immunohistochemical expression such as diffuse and strong reactivity for synaptophysin and NSE, along with obvious non-expression for GFAP. Strong immunostaining for synaptophysin has been recognized as the most reliable diagnostic marker [4, 7]. Typically, synaptophysin immunoreactivity is noted in the neuropil, especially in fibrillary zones and perivascular cell-free areas, and not in the cell bodies of normal neurons [4]. Immunostaining for neuron-specific enolase (NSE) also determined the neuronal nature of the neoplasm. The significance of GFAP staining herein is to identify the presence of glial cell differentiation; thus, it is used to exclude diagnoses of oligodendroglioma, astrocytomas, and ependymoma. Although most studies have shown that GFAP is expressed only in entrapped reactive astrocytes, it has also been reported in CN cells by some authors [8, 9].
Because of the fore-mentioned overlapping histological appearances, there is a wide variety of differential diagnoses of CN, in which the two most common are oligodendroglioma and ependymoma. Oligodendroglial tumors and CN can have cytoplasmic vacuoles and monomorphic histologic appearances, which can hardly be differentiated from each other [9]. However, intraventricular localization, along with histological features of anuclear neuropil islands, salt and pepper chromatin, eosinophilic granular bodies, and lack of infiltrated brain parenchyma may suggest a diagnosis of central neurocytoma [9]. Immunoreactivity for synaptophysin and NSE may support the diagnosis. An intraventricular tumor with a solid, non-infiltrating pattern along with perivascular pseudo-rosettes is likely a feature of an ependymoma rather than CN [9, 10]. Nevertheless, characteristics of nuclear roundness with speckled chromatin and comparatively poorly-defined perivascular rosettes suggest neurotic origin, which is confirmed by synaptophysin immunoreactivity along with no expression of GFAP [6].
Regarding prognosis, CN is generally benign, with a cure of an extended resection. Overall survival rate at 3 and 5 years after gross total resection was 95% and 85%, respectively, versus 55% and 45% after subtotal resection [11]. In one multicenter study of 71 patients, incomplete resection was predictive of poor outcome [12]. The patient presenting tumor hemorrhage and supratentorial ventricular system dilation, cerebellum, and brainstem compression along with uncomplete tumor resection may deal with unfavorable outcomes.
Conclusion
Central neurocytoma, a kind of neurocyte tumor, occurs mostly in the lateral and third ventricle and usually affects young adults. The neoplasm shares some similar radiological features, histologic appearance with oligodendroglioma or ependymoma, and shows specific neuronal immunoreactivity. Due to rare incidence, CN is probably misinterpreted with other tumors such as oligodendrogliomas and ependymoma. From our practical case and review of the literature, imaging and clinical symptoms provide an approach for initial diagnosis of CN; however, histological and immunohistochemical appearance shows excellent importance for diagnosis confirmation.