






1. INTRODUCTION
Rasmussen encephalitis or chronic focal encephalitis is a rare disease of childhood. The disorder is characterized by refractory seizures, progressive neurological deficits and cognitive impairment [1]. The exact aetiology is still not established. The condition usually presents as pharmacoresistant seizures. The most useful investigations are EEG and serial MRI brain but brain biopsy remains the gold standard for establishing the diagnosis [2]. Despite the advances in medical treatment, surgery remains the only cure of disease progression but not without neurological deficit [3]. Here we present a case of Rasmussen encephalitis followed by viral encephalitis in a child.
2. CASE PRESENTATION
A 8 year old male child presented in emergency department of Nishtar Hospital Multan. His chief complaints were high grade fever for 5 days and fits for 3 days. Fits were characterized by stiffening followed by jerking movements of whole body associated with frothing from mouth, 2-3 episodes per day each lasting for 8-10 minutes. There was no history of headache, rash, loss of consciousness, change in bowel habits, respiratory or urinary symptoms.
At the time of presentation patient was hemodynamically unstable with pulse rate of 130 beats per minute, blood pressure of 100/60 mmHg and an oral temperature of 39.4oC. Signs of meningeal irritation were positive. Reflexes were brisk with extensor planter on both sides. The rest of systemic examination was unremarkable. The patient was diagnosed with viral encephalitis on the basis of clinical features and CSF examination. Patient had uneventful recovery and was discharged after 2 weeks.
After 2 years patient again presented in emergency department with history of fits and progressive weakness of left half of body for three months. Fits were initially focal involving face, 4-5 episodes per day each lasting for 2-3 minutes. Later, he developed left sided tonic fits with twitching of face which lasted for 4-5 minutes. The frequency of fits increased over time and for the last 5 days increased to 8-10 episodes per day. He had no complaint of fever, headache, change in bowel habits, respiratory or urinary symptoms.
Clinical examination revealed a conscious oriented child of average built with slurred speech. Cranial nerves examination was normal. Examination of motor system revealed increased tone, power of 4/5, and brisk reflexes with extensor planter on left side. Examination was normal on right side. Signs of meningeal irritation were negative. There were no involuntary movements or cerebellar signs. The rest of systemic examination was unremarkable.
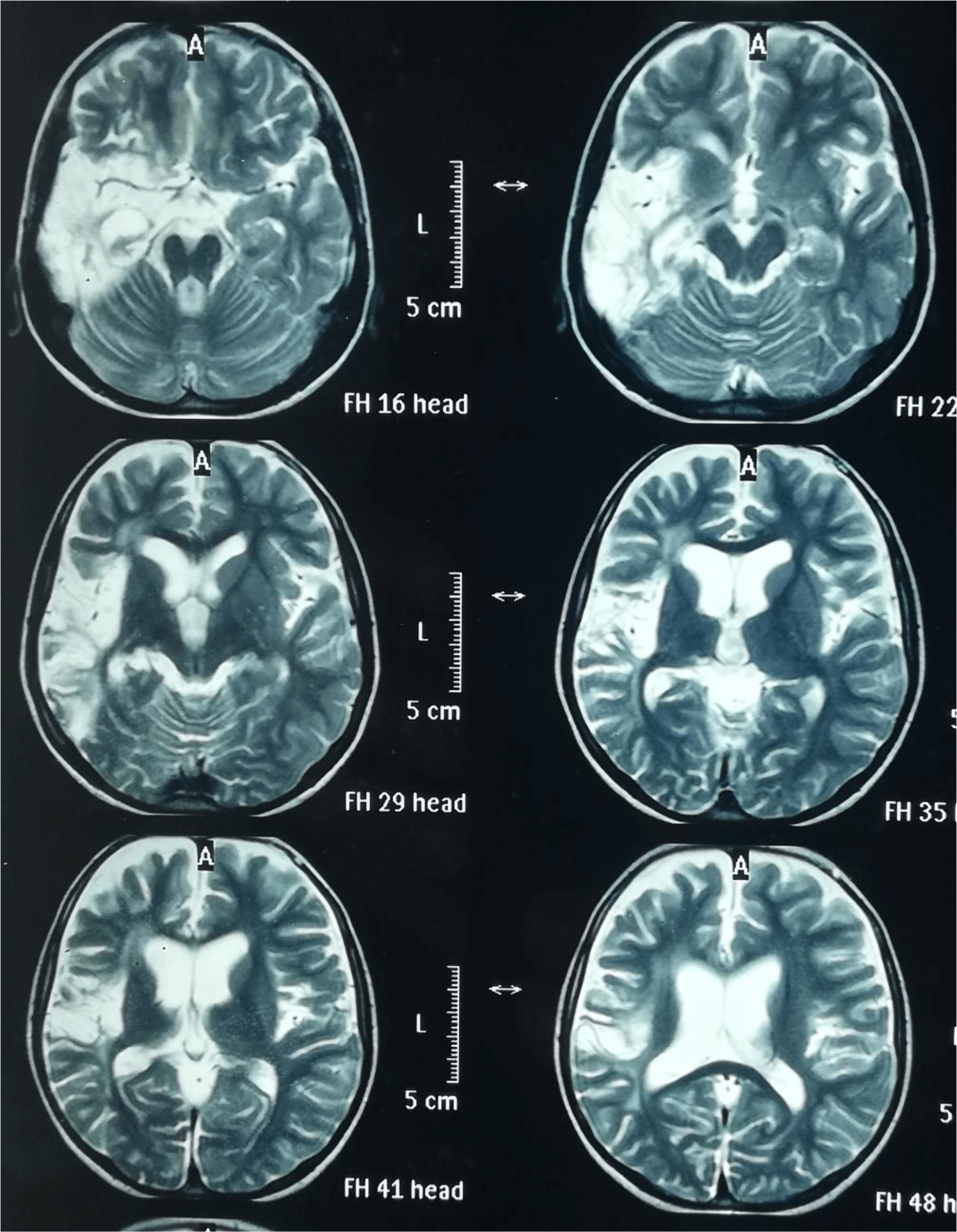
MRI brain was ordered which showed abnormal focal post contrast enhancement along right temporoparietal region with exvaco dilatation of left lateral ventricle suggestive of unilateral cortical atrophy with minimal surrounding areas of gliosis (Figure 1). EEG was also ordered which showed generalized epileptogenic activity over slow background. Brain biopsy was not performed in this patient. The parents were counselled about the significance of brain biopsy but they refused. On the basis of clinical and radiological evidence, diagnosis of Rasmussen encephalitis was made. Corticosteroids (1 mg/kg/day), valproic acid (15 mg/kg/day), carbamazepine (10 mg/kg/day) and levetiracetam (20 mg/kg/day) were prescribed which reduced the frequency of seizures over a period of 3 months.
3. DISCUSSION
Rasmussen encephalitis is a rare and progressive disorder of central nervous system usually affecting one brain hemisphere. It usually presents in children 6-8 years old although adolescents and adults can be affected. Around 10% of all cases are adolescents and adults [3].
Rasmussen encephalitis is characterized by brain inflammation resulting in unilateral brain atrophy; clinically by intractable seizures, progressive neurological deficits and cognitive impairment including language skills if dominant hemisphere is involved [1]. The exact aetiology is still unknown but it is sporadic with no genetic link [3]. Multiple theories are proposed including the presence of neuronal antibodies and T cell mediated mechanisms triggered by viral infection [4]. It is proposed that neurons expressing autoantigen or foreign antigen are attacked by Granzyme B positive cytotoxic T cells. The neurons under cytotoxic T cell attack are injured and later engulfed by microglia and macrophages which results in progressive brain tissue loss.
The most common presentation is pharmacoresistant seizures. Seizures may have different forms but most notable seizure manifestation is epilepsia partialis continua which is continuous twitching of face, arm or leg on one side of body. About 56-92% of patients with Rasmussen encephalitis have epilepsia partialis continua [3].
Encephalitis occurs in three stages, first prodromal stage consisting of infrequent seizures and mild hemiparesis; this is followed by acute stage leading to frequent pharmacoresistant seizures, hemiparesis, hemianopia and dysphasia if dominant hemisphere is involved. Finally there is chronic stage with less seizure activity and fixed neurological deficit [5].
Diagnosis is made based on typical clinical features and investigations. The most useful investigations are MRI brain: which shows progressive atrophy and scarring of one side of brain, EEG: which may reveal electrical features of epilepsy. Brain biopsy is the gold standard investigation but it is not usually performed. Brain biopsy may reveal perivascular T-lymphocytic infiltration, microglial proliferation, several microglial nodules and evidence of neuronophagia [2].
Various treatment strategies have been proposed for example antiepileptic drugs, immunosuppressive or immunomodulatory therapies and surgical treatment. Antiepileptic drugs are frequently helpful but usually do not entirely control seizures. Corticosteroids, IVIG or plasma exchange can improve symptoms in early stage of disease. Immunosuppressive or immunomodulatory therapies have shown some success in improving symptoms but failed to halt the progress of disease in long term. Recently monoclonal antibody against tumor necrosis factor-α (adalimumab) has been found to control seizures and preserve cognitive function in approximately 50 % of patients [6].
The most effective treatment to cure disease progression is surgery. Surgical procedures such as anatomical hemispherectomy (surgery to remove the affected hemisphere) and functional hemispherotomy (surgically severing all connections between right and left halves of brain) may control seizures and also improve behaviour and cognitive abilities [3].
Our patient presented with typical clinical features of Rasmussen encephalitis. The diagnosis was made on the basis of clinical judgement, radiological evidence of unilateral brain atrophy and EEG showing epileptogenic activity. We treated our patient with conventional medical therapies which reduced the frequency of seizures. The option of hemispherectomy was discussed with parents and was refused.
Conclusion
Our patient presented with clinical history of Rasmussen encephalitis. Though we diagnosed our patient on the basis of strong clinical suspicion and imaging studies, we tried available conventional therapies to treat our patient at our best. The disorder is refractory to conventional medical treatment with surgery being the most effective treatment.