






1. INTRODUCTION
Pheochromocytomas are tumors arising from the catecholamine-producing cells in the adrenal medulla. The typical symptoms of pheochromocytoma include headache, sweating, tremor, pallor and palpitations. Due to excessive paroxysmal catecholamine secretion into the bloodstream, the symptoms are usually episodic and associated with considerable cardiovascular complications and even death [1-3]. Previously, only 10% of pheochromocytoma cases were thought to be hereditary as proposed by Emanuel Bravo in 1984 [4]. Recently, with the development of genetic technologies, germline mutations have been found to contribute up to 30–40% of the pheochromocytoma (and paraganglioma) cases [5-8]. Genetic testing for all cases of pheochromocytoma/paraganglioma are now recommended as part of the standard care [6, 9-11]. In addition to guiding patient management, genetic results also provide clinicians with the ability to screen for pheochromocytoma and related tumors in relatives of a mutation-positive proband [12-14]. However, genetic testing is not routinely performed in Vietnam due to the limitation of technologies and resources. Here we report the first pheochromocytoma cases that were referred to our center for genetic testing and how the results have impacted clinical practice.
2. MATERIALS AND METHOD
This study was approved by the Ethical Committee of The University of Medicine and Pharmacy at Ho Chi Minh City, Vietnam. Patients were admitted to Cho Ray Hospital and University Medical Center between 2016 and 2018. Pheochromocytomas were diagnosed based on the elevation of 24-hour urinary catecholamine and plasma free metanephrine, computed tomography (CT) imaging, and postoperative pathology. Other clinical information including age, gender, syndromic characteristics of multiple endocrine neoplasia type 2, neurofibromatosis type 1, Von Hippel-Lindau disease, familial history of pheochromocytoma, medullary thyroid carcinoma, head and neck paraganglioma, serum calcium, parathyroid hormone, and calcitonin were also recorded. Patients were counseled and provided written informed consent for genetic testing. If a germline mutation was found, all first-degree relatives of mutation-positive patients were also counseled and provided written informed consent for genetic testing. If genetic testing was indicated in the children, the written informed consent was obtained from their next of kin, caretakers, or guardians.
All the results of biochemical diagnostic tests in this study were performed in the standard biochemical laboratory of either Cho Ray hospital or the University Medical Center following the criteria of ISO 15189:2012.
GeneJET Whole Blood Genomic DNA Purification Mini Kit (Thermo Fisher Scientific, Massachusetts, USA) was used to extract the genomic DNA of the subjects according to the manufacturer’s protocol and these samples were stored at - 20°C for further experiments.
Genetic testing for pheochromocytoma patients was performed following the American Endocrine Society decisional algorithm [6]. All coding exons and flanking regions of VHL (GenBank NG_008212.3), SDHB (GenBank NG_012340.1), SDHC (GenBank NG_012767.1), SDHD (GenBank NG_012337.3), MAX (GenBank NG_029830.1),
TMEM127 (GenBank NG_027695.1) genes and RET (GenBank NG_007489.1) exons 10, 11, 13, 14, 15, 16 were amplified and sequenced. DNA amplification was performed by Mastercycler@proS, the results of DNA electrophoresis on 1% agarose gels with Diamond™ Nucleic Acid Dye (Promega, Madison, WI, USA) were used to confirm the appropriate length of amplified products. These products were subsequently purified with Exosap-IT glycerol solution (Thermo Fisher Scientific, Massachusetts, USA) and then sequenced by BigDye® Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA). Sequencing reactions were analyzed with ABI 3130 Genetic Analyzer (Applied Biosystems). Results were compared to the reference sequences of target genes in Genebank. All the primers for genetic testing are listed in Supplementary Table 1.
3. RESULTS
All patients (4 females and 3 males) referred to our center were at high risk of germline mutation as they were diagnosed with bilateral pheochromocytoma and/or at early age of onset, and/or had a familial history of either pheochromocytoma or medullary thyroid cancer. Two patients BN01 (with unilateral pheochromocytoma) and BN02 (with bilateral pheochromocytoma) were tested for VHL, SDHB, SDHC, SDHD, MAX, TMEM127, and RET mutations but none were detected. Multiple endocrine neoplasia type 2A (MEN2A) manifestations were found in 3 out of 7 cases (BN05, BN06, BN07), the other patients were diagnosed with pheochromocytoma only. None of the probands showed manifestations or family history of Neurofibromatosis type 1, von-Hippel-Lindau syndrome or paraganglioma. Four of seven patients (BN02, BN03, BN04, and BN07) were diagnosed with bilateral pheochromocytoma, whilst BN05 and BN06 developed another pheochromocytoma after the first adrenalectomy surgery. Clinical characteristics of these patients are summarized in Table 1. Family history of these patients also showed that BN03, BN04 and BN05 had siblings who developed either pheochromocytoma or medullary thyroid cancer and the deaths of these family members were described as sudden and at a very young age (from 30s to 40s years old).
In patients with MEN2A syndromic manifestations, RET gene sequencing was indicated. On the other hand, the order of genetic testing in patients with only signs and symptoms of pheochromocytoma were performed according to the Endocrine Society decisional algorithm6. RET gene mutations were found in 3 out of 3 MEN2A patients (BN05, BN06, and BN07) and in two of three patients with bilateral pheochromocytoma (BN03 and BN04). All RET mutations were in codon 634 and either c.1900T>C or c.1900T>G (Table 1).
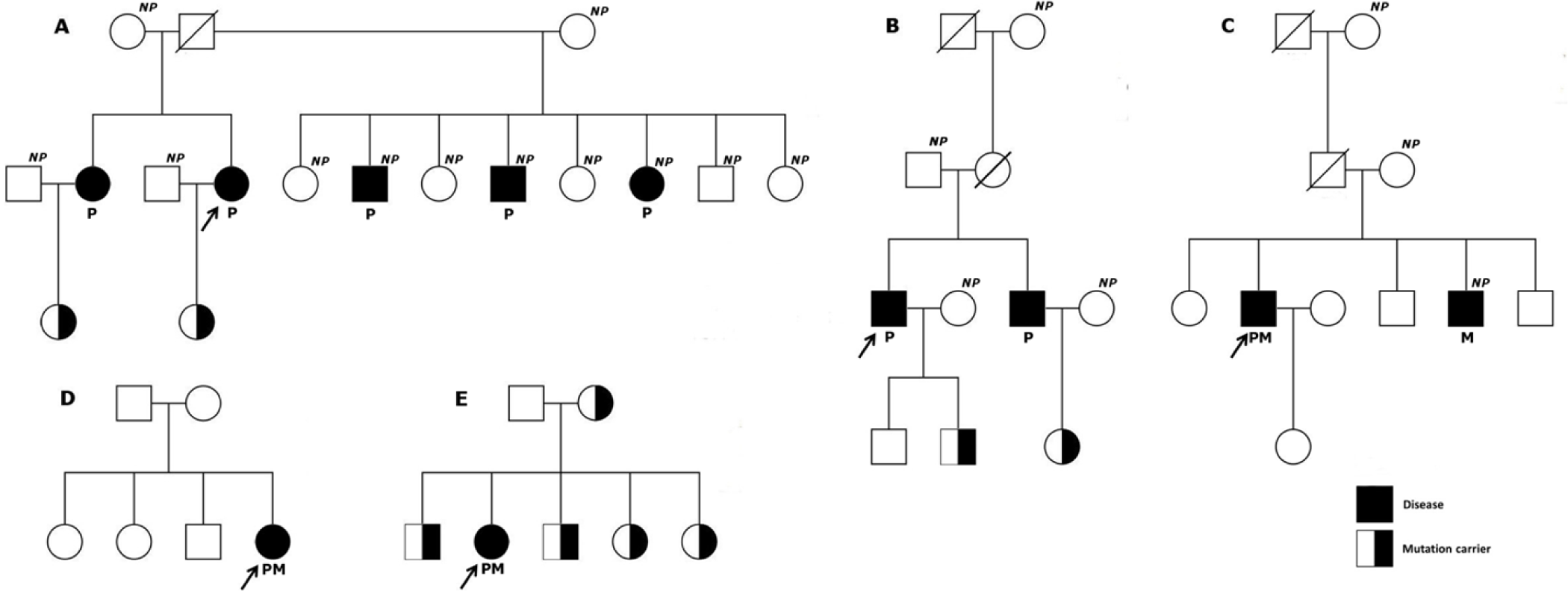
Genetic analysis in the first degree relatives of mutation-positive probands showed an inherited pattern in all the families except the family of BN05 and BN06. However, patient BN05 presented with typical MEN2A phenotype and medullary thyroid cancer was reported in one of his siblings (who declined genetic testing) suggesting that the disease in this family was highly likely inherited. Interestingly, genetic testing in patient BN06 suggested that this patient carried a ‘de novo’ mutation as none of the first degree family members carried this RET mutation. Currently, the screening for disease has been undertaken periodically for mutation carriers without any phenotypic signs and symptoms. The pedigrees of mutation-positive probands and their relatives’ mutation status are illustrated in Figure 1.
4. DISCUSSION
Despite the fact that germline mutations may account for up to a total of 30-40% pheochromocytoma/paraganglioma cases and this disease can beheritable [5, 15], genetic testing for pheochromocytoma is still not routinely performed in Vietnamese patients. In this study, we found that five of six patients with bilateral pheochromocytoma carried germline mutation in codon 634 of RET proto-oncogene and four of five mutation-positive probands showed increased plasma adrenaline and noradrenaline.
We also reported a potential ‘de novo’ mutation of RET in patient BN06. It has been reported that ‘de novo’ RET mutations have been identified in as many as 9% of MEN2A and medullary thyroid carcinoma (MTC) cases and this once again emphasizes the role of genetic testing in patients who are diagnosed with pheochromocytoma/MTC without any apparent family history [16]. It should be noted that up to 7.3% of RET germline mutation carriers have no family history [17, 18]. Knowledge regarding the genetic background of patients with pheochromocytoma has the potential to improve management by providing a more accurate risk stratification and follow-up plan as well as genetic counseling and prophylactic thyroid surgery [14, 19]. For example in the case of patient BN04, after RET mutation identification, the patient was referred for thyroid examination. Thyroid ultrasound described a hypoechoic nodule 4x7 mm and the result of fine needle aspiration was ‘normal’. The patient agreed to undergo prophylactic total thyroidectomy and the pathology result of the thyroid nodule showed medullary thyroid cancer.
It is very important to understand the genetic status of first-degree relatives of mutation-positive patients. Once specific mutations are found in the relatives, it will totally change the medical strategies for screening and monitoring of related tumors14. In patients BN04 and BN07 family; genetic counseling, monitoring plan, and prophylactic thyroid surgery were offered to all the patients’ family members who carried the RET mutation. Even if the prophylactic thyroidectomy is not performed, physical examination, ultrasound, and biochemical monotoring annually are very useful for these subjects.
This study has several limitations that need to be discussed. First, the sample size is small due to the fact that pheochromocytoma is not a prevalent disease and only patients with a high risk for having genetic mutation were recruited. This selection bias may overemphasize the benefits of genetic testing. Second, because of the disadvantage of direct sequencing technique, neither SDHA, SDHAF2 or other PC/PGL candidate genes were included in the genetic testing panel. Advanced sequencing technique which was proven competent for long and complex genes can be applied in the future to better detect genetic mutations in pheochromocytoma [20-22].
Current genetic testing algorithms are based either on the 2014 American College of Medical Genetics or the 2014 Endocrine Society Clinical Practice Guidelines [6, 9]. Both guidelines recommend that all patients with pheochromocytoma should consider genetic testing as part of their management. However, to our knowledge, this is the first report of genetic testing in a series of Vietnamese pheochromocytoma patients in Vietnam and we expect that this result will leverage the role of genetic testing of pheochromocytoma patients to overcome barriers such as high cost and social-emotional concern.